摘要
裘馨氏肌肉萎縮症 (Duchenne muscular dystrophy, DMD) 為我國罕見疾病之一,據北部大型醫學新生兒篩檢中心統計,發生率約1/7000的活產男嬰,治療上以美國 FDA 提供的治療準則為主,利用類固醇控制肌肉萎縮情況。近年來有數種創新的研究與治療方法,包含(1)通讀轉錄 (Read through) 治療移碼突變 (frameshift mutation),(2)反義寡核甘酸 (antisense oligonucleotides, ASOs) 治療特定外顯子突變 (exon mutation),(3)adeno-associated virus, AAV 病毒載體基因治療。目前核准上市的藥物包含 Translarna (Ataluren) 與 Exondys 51 (Eteplirsen),對裘馨氏肌肉萎縮症患者的症狀改善有一大進展。對於 DMD 的病理機轉仍有很多嶄新的治療方法值得研究探討,本文回顧近來研究,提供對藥物治療方法與策略。
關鍵字:裘馨氏肌肉萎縮症、肌肉失養蛋白、基因治療、Duchenne muscular dystrophy、dystrophin、gene therapy
壹、簡介
裘馨氏肌肉萎縮症 (DMD) 是一種主要影響男性肌肉病變的X染色體隱性遺傳疾病,情況較為輕度者,通常是基因缺失的帶原者或缺失的位置不甚重要,另稱為貝克氏肌肉萎縮症 (Becker muscular dystrophy, BMD)(圖一)。
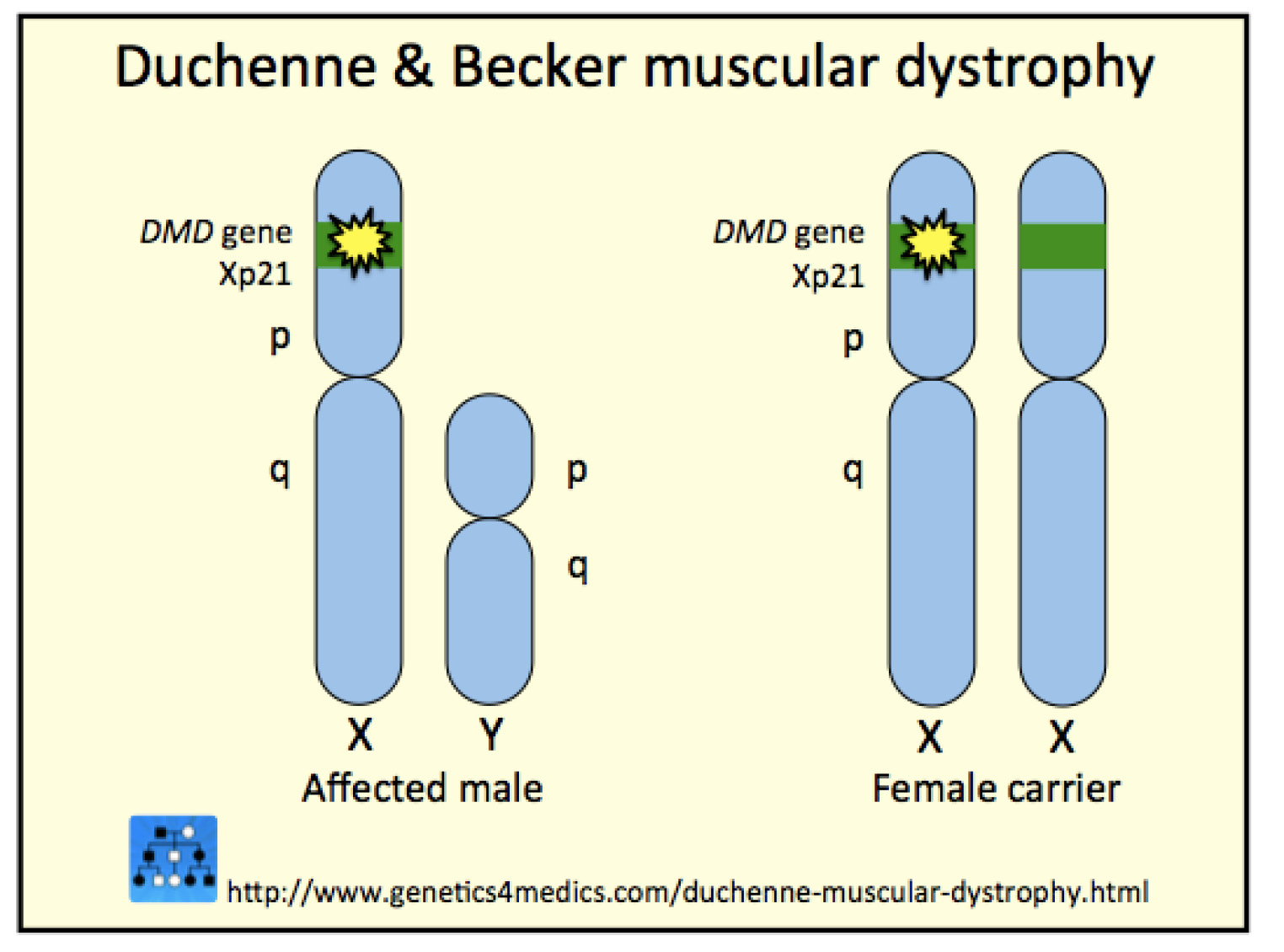
圖一 肌肉萎縮症基因缺失位置
1852年 DMD 相關臨床症狀首度由法國醫師 Duchenne 發表後,歷經一個多世紀以來都沒有進展,直至近代分子醫學的進步,才於1987年發現 DMD 患者在肌肉失養蛋白 (dystrophin) 有異常的表現,患者肌膜容易破裂,導致血清中肌酸激脢 (creatin kinase) 的濃度異常升高。分析 DMD 患者的基因表現,可在 Xp21.2的位置上發現缺失 (約70%) 或重複 (約8%),據新生兒篩檢中心統計,台灣活產男嬰的發生率約1/7000,依衛福部 ICD-10-CM 公告 DMD 為我國罕見疾病 (G71.0)1。
貳、藥物治療
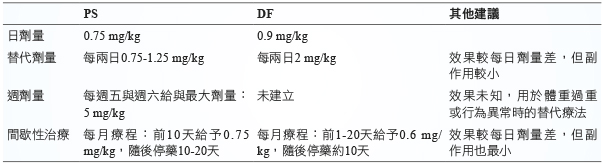
目前針對 DMD 的治療並沒有痊癒的方法,僅能藥物控制病程的惡化,並以類固醇為主,常用的藥物包含 prednisone (PS) 與 deflazacort (DF),使用上將預期能達到下列幾個治療目標:一、增加肌肉強度與活動時間,二、增進肺臟呼吸功能,三、減少不必要的外科手術,四、延緩後期心臟衰竭的時程。使用 prednisone (PS) 與 deflazacort (DF) 兩種類固醇在初期肌肉改善上,並沒有明顯差異,但 DMD 患者後期引起的肺臟或心臟衰竭,則沒有兩種類固醇治療上的相關比較。兩者劑量上的使用比較整理 (表ㄧ)2-3。
表一 prednisone (PS) 與 deflazacort (DF) 治療 DMD 的比較
研究臨床藥物用於治療的原則,大致可分為三個方向:一、通讀轉錄 (Read through) 治療移碼突變 (frameshift mutation),二、利用反義寡核甘酸 (antisense oligonucleotides, ASOs) 跳過特定外顯子突變 (exon mutation),三、利用 AAV 載體重新修飾無功能或部分功能的 dystrophin 蛋白,製造部分具功能的 dystrophin 蛋白4。
一、通讀轉錄 (read through) 治療,當 DMD 發生移碼突變 (frameshift mutation),通常於外顯子51最容易發生突變,使該基因點位開始的下游蛋白質讀碼框移轉而無法製造完整的蛋白,FDA 已於2016年9月核准上市的藥物 eteplirsen 為一小片段基因聚合物 (oligomer),可用於修飾轉錄失養蛋白的前訊息 RNA (pre-mRNA),修飾移碼突變基因而重啟停止的序列,繼續轉錄下游蛋白,製造出部分具功能性的肌肉失養蛋白5。
二、利用反義寡核甘酸 (antisense oligonucleotides, ASOs) 跳過特定外顯子突變 (exon mutation),製造部分具功能的 dystrophin 蛋白。正常基因在 DNA 轉錄 mRNA 後 (圖二a),方能進行後續轉譯蛋白質的工作,由英國牛津大學藉基因檢測將 DMD exon 50 缺失的病人利用反義寡核甘酸 (Anti-sense oligonucleotides, ASOs) 進行治療,原 exon 50 基因缺失後,會造成基因下游無法被轉譯,生成無功能的蛋白質 (圖二b),經過設計的 ASOs 作用於 DMD pre-mRNA 上的 exon 51,避免 mRNA 剪接調控蛋白 (mRNA splicing regulatory proteins) 遭遇遺失的 exon 後終止,使 exon 49與 exon 51連結後恢復下游基因的轉譯作用,製造出雖被截短,但仍具部分功能的 dystrophin,恢復部分肌肉作用 (圖二c)6,減少肌肉萎縮情形。
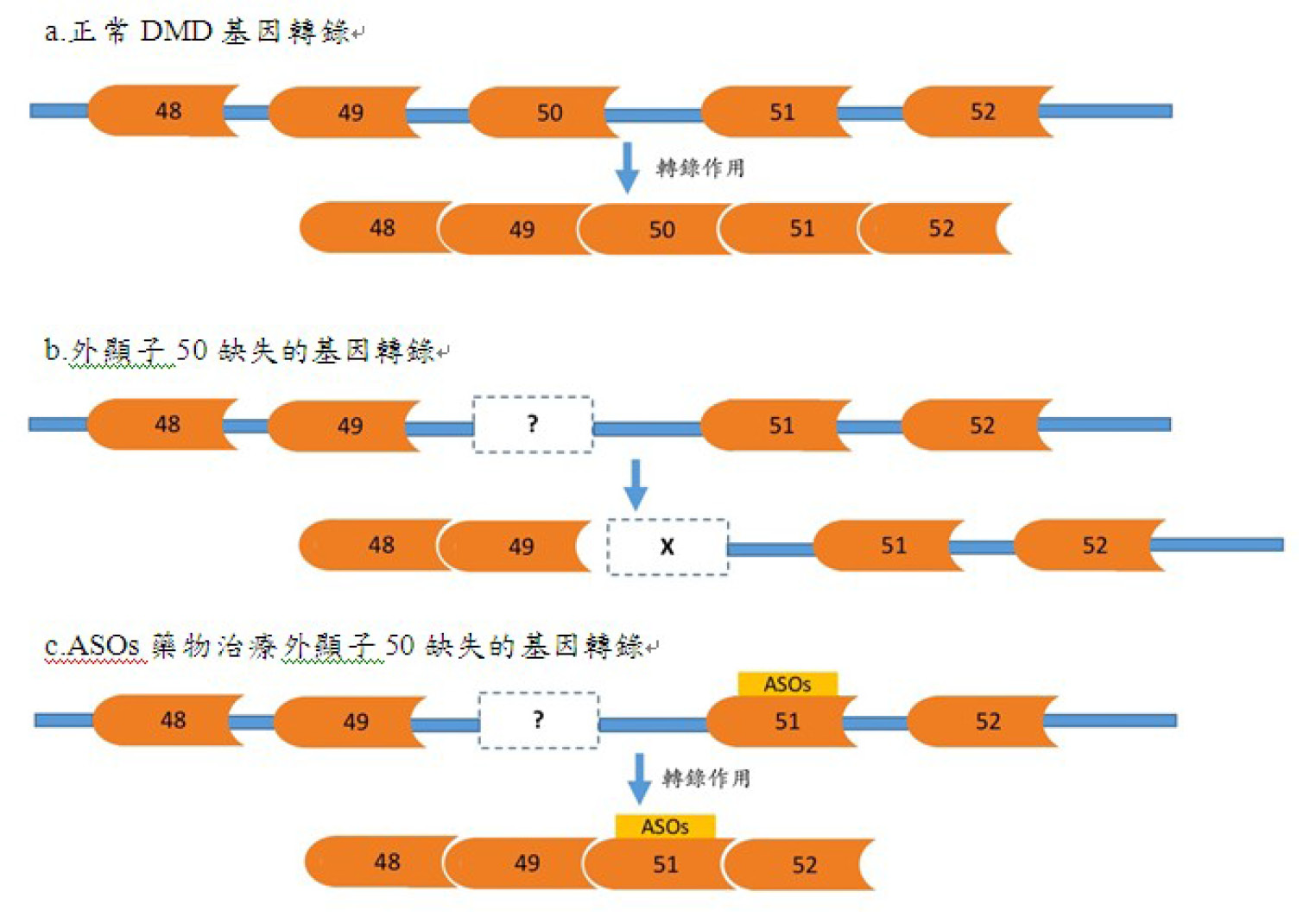
圖二 ASOs 藥物作用於 DMD 基因缺失的治療
據萊登大學醫學中心 DMD資 料庫統計,約有83%的 DMD 病人適合利用 ASOs 的原理進行治療,若 DMD 突變超過36個 exons 以上的大缺失,則不適合以 ASOs 治療,萊登大學持續進行 phase III 臨床試驗,利用 ASOs 類的試驗藥物2-O-methyl phosphorothioate (2OMe-PS) 進行4名男孩的 DMD 治療,基因缺失的位置分別位於 DMD exon 50、exons 48-50、exons 49-50、與 exon 52,以肌肉注射的方式給藥,利用藥物修飾 exon 51的結構後,使剪接調控蛋白利用修飾後的 exon51與上游或下游基因重新連結,製造出不完整但可持續轉譯的片段 mRNA,製造出下游蛋白,給藥四週後,由實驗結果觀察4名男童注射部位,製造出具有部分功能的 dystrophin,且將其中1名男孩進行肌肉切片,有64%-97%肌纖維上的肌漿膜 (sarcolemma) 都有功能性的 dystrophin,經染色比對健康肌細胞後,更有17%-35%肌肉強度6-8。近年許多公司積極投入研發此類治療,而 Sarepta Therapeutics 研發的 EXONDYS 51 (eteplirsen) 於2016年9月剛獲美國 FDA 核准上市。
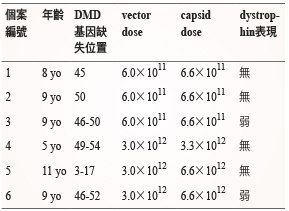
三、藉由載體的基因治療 (Vector-mediated gene therapy),針對 DMD 基因遺失片段的取代治療,利用 AAV 病毒作為載體,攜帶此段遺失的 DMD 基因 mini-dystrophin,局部注射至肌肉。AAV 是容易感染肌肉細胞的病毒,在動物模式的研究中成功的取代遺失的基因片段,可合成 dystrophin 蛋白並正常運作。此治療方式接著進行 Phase I 臨床試驗,採隨機雙盲對照實驗,選擇6名 DMD 的男童 (5-11歲),將載體局部注射至單側肱二頭肌,進行為期1-2個月追蹤觀察,嚴密監測各項免疫反應,結果統計後 (表二),在肌肉失養蛋白的表現上卻沒有觀察到良好的結果9。
表二 臨床試驗第一期利用 AAV 治療 DMD 結果9
藥物運用於基因的治療研究持續進行,結果有符合期待,也有令人失望的結果,DMD 基因突變頻率最高的位置為 exon51,在此部位進行相關臨床試驗的研究較多,倘若缺失的部分為其他基因位置,則侷限了這些研究藥物的效能。此外,臨床試驗的治療上對於沒有注射藥物的區域,則沒有改善,對於肌肉功能無法全面性的修復。綜合上述,目前 DMD 治療效果仍有限,僅對於部分 DMD 病人有嶄新的治療突破。文獻上已發展較多的 DMD 治療機制主要為透過 read through,修復 stop codons 而製造部分功能失養蛋白,在2014年7月上市的 Translarna (Ataluren) 獲歐盟委員會有條件批准 (conditional approval) 用於具行動能力的5歲以上 nonsense mutation DMD 患者口服治療,未來的研究仍可朝更多創新方向著手。
The Treatment Nowadays in Duchenne Muscular Dystrophy
Wei-Chih Chen1, Pei-Ling Liao1, Pei-Chun Kuo1, Yung-Hung Hsieh2, Ni-chung Lee3
Department of Pharmacy Practice, Tri-Service General Hospital1
Department of Pharmacy, Taichung Armed Forces General Hospital2
Department of Medical Genetics, National Taiwan University Hospital3
Abstract
Duchenne muscular dystrophy, DMD, is a rare disease in Taiwan, ROC. According to the database of NTUH Newborn Screening Center, the prevalence is about 1 in 7000 male newborns. The treatment guideline is based on the FDA suggestion, the corticosteroid is useful in the control of muscular dystrophy. Recently there are many innovative therapeutic approaches underways, including (1) read through treatment for DMD frameshift mutation, (2) antisense oligonucleotides in specific exon mutation, (3) adeno-associated virus as vectors in gene therapy. All the researches are helpful in management of the DMD patients. The pathological mechanisms are still worth of research. We review recent articles for more development new treatment methods.
參考資料:
1. Wei-Chih Chen, Ni-Chung Lee, Wuh-Liang Hwu, Detection of the DMD gene deletion using DNA extracted from dried blood spot. 2015: Molecular medicine, College of Medicine, National Taiwan Univerity Hospital. p. 1-8.
2. Rao, V.K., Guidelines for Corticosteroid use in Treatment of DMD. Pediatric Neurology Briefs, 2016. 86(5): p. 465-472.
3. Katharine Bushby, David J Birnkrant, Laura E Case, et al: Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. The lancet neurology, 2010. 9: p. 77-93.
4. Yuko Shimizu-Motohashi, Hirofumi Komaki, Shin’ichi Takeda, et al: Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. American Journal of Translational Research, 2016. 8(6): p. 2471-2489.
5. Hong M. Moulton, Jon D. Moulton, Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchene muscular dystrophy, Biochimica et Biophysica Acta, 2010. 1798: p. 2296-2303
6. Wilton SD, V.R., Fletcher S, The emperor’s new dystrophin: finding sense in the noise. Trends in molecular medicine, 2015. 21(7): p. 417-426.
7. Aartsma-Rus, Fokkema IF, Van Ommen GJ, et al: Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle & Nerve, 2006. 34(2): p. 135-144.
8. Aartsma-Rus, Verschuuren J, Ginjaar I, et al: Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human mutation, 2009. 30(3): p. 293-299.
9. Dawn E Bowles, Chengwen Li, Steven J Gray, et al: Phase 1 Gene Therapy for Duchenne Muscular Dystrophy Using a Translational Optimized AAV Vector. The American Society of Gene & Cell Therapy, 2012. 20: p. 44-455.

