摘要
修格蘭氏症候群為一進展性自體免疫疾病,特徵為淋巴球浸潤破壞腺體造成乾燥症狀,目前自體免疫上皮炎是其致病機轉的主流理論,即上皮細胞成為一非典型之抗原呈現細胞,持續大量表現 TLR3受體使上皮細胞成為自體免疫反應的調節中心。由於致病機轉逐漸明瞭與臨床試驗需求,2016年版之 ACR/EULAR 分類準則提出,除了現行使用的藥物 (pilocarpine 與 cevimeline 等) 外,將有更多的生物製劑得以使用於治療修格蘭氏症候群。
關鍵字: 修格蘭氏症候群、Sjögren's syndrome、乾燥症狀、2016 ACR/ EULAR
壹、前言
口乾眼澀的夢靨,乾苦一生何人知!修格蘭氏症候群 (Sjögren's syndrome,SS) 為一進展性自體免疫疾病,由醫師 Hendrik Sjögren 發現而後得其名,特徵為因淋巴球浸潤破壞唾腺 (salivary gland) 及淚腺 (lacrimal gland) 造成乾燥之症狀 (sicca symptoms)1。
修格蘭氏症候群可細分為原發性及次發性兩類型,原發性修格蘭氏症候群 (primary Sjögren's syndrome,pSS) 與次發性修格蘭氏症候群 (secondary Sjögren's syndrome) 之臨床特徵相仿,治療方式亦雷同,差別僅在於次發性修格蘭氏症候群用於描述該疾病同時伴隨著其他全身性自體免疫疾病 (如:類風濕性關節炎、全身性硬化症或全身性紅斑性狼瘡) 者1,2。
修格蘭氏症候群之盛行率受研究設計與分類標準影響而變異性大,其盛行率約為0.01%-0.72%,以女性為主 (女男比約10:1),任何年齡皆可能患病,但30-50歲居多,種族特異性尚不甚明確,目前僅有一研究指出非歐裔之盛行率高於歐裔約兩倍2。
貳、 修格蘭氏症候群的病生理學致病機轉
多種因素及個體基因皆被認為影響著修格蘭氏症候群的進展2,以下就基因、免疫反應及其他因素分述如下:
一、基因
藉由全基因組關聯研究 (genome-wide association studies;GWAS) 技術,可以確定許多與修格蘭氏症候群相關的基因,例如:位於人類第6對染色體長臂21位置 (6q21) 上的人類白血球抗原 (human leukocyte antigens;HLA)-DQB1區域,負責編碼人類的主要組織相容性複合體 (major histocompatibility complex;MHC) 分子,被認為與修格蘭氏症候群高度相關3,4。
另外,與干擾素調節因子5 (Interferon regulatory factor 5) 有關的基因 IRF5、訊息傳遞轉錄活化蛋白4 (signal transducer and activator of transcription) 相關之基因 STAT4、負責編碼B淋巴球激酶 (B lymphocyte kinase) 的 BLK、編碼 IL-12次單元α(IL-12 subunit α) 的 IL12A、編碼 TNFAIP3交互作用蛋白 (TNFAIP3-interacting protein 1) 之 TNIP1及 CXC 趨化因子受體5 (CXC chemokine receptor 5) 關聯之基因 CXCR5亦與修格蘭氏症候群有關3,4。
二、免疫反應
自體免疫上皮炎 (autoimmune epithelitis) 是目前修格蘭氏症候群致病機轉的主流理論,上皮細胞 (epithelial cells) 成為一非典型之抗原呈現細胞 (atypical antigen-presenting cells),持續大量表現免疫活性分子 (immune-competent molecules),使上皮細胞成為自體免疫反應的調節中心為本理論的核心2。
修格蘭氏症候群患者之唾腺或淚腺上皮細胞會持續表現 TLR3受體 (Toll-like receptor 3),此現象可能由基因因素或病毒感染造成,TLR3受體會經由 TRIF (TIR-domain-containing adapter-inducing interferon-β) 轉接蛋白與 IRF3 (interferon response factor 3) 蛋白活化下游途徑調控干擾素β(interferon beta,IFNβ) 等細胞激素 (cytokine),干擾素β可活化T細胞進一步藉由釋出白血球間素-17(interlukin-17,IL-17) 與干擾素 γ(IFNγ) 等細胞激素傷害組織,干擾素β亦藉由B細胞活化因子 (B cell activating factor,BAFF) 活化B細胞並造成組織的傷害2,5,此現象合理解釋於修格蘭氏症候群患者之唾腺有B細胞活化因子升高的現象3,另外,T細胞亦可藉細胞激素調控B細胞,形成T細胞與B細胞相關性之連結,前述的組織傷害會造成腺體功能下降,形成乾燥症狀 (圖一)2,5。TLR3受體也會經由 TRIF 轉接蛋白活化下游的凋亡蛋白酶3 (caspase 3) 啟動細胞凋亡路徑6,造成上皮細胞的凋亡,形成腺體分泌功能障礙2。
另外,TLR3訊息傳遞路徑亦可使修格蘭氏症候群A自體抗原 (Ro/SSA autoantigens) 與修格蘭氏症候群B自體抗原 (La/SSB autoantigens) 等自體抗原由唾腺或淚腺上皮細胞之細胞質移動到細胞膜上進行大量表現;而上皮細胞凋亡時,也可使含有 Ro/SSA 自體抗原及 La/SSB 自體抗原之小泡 (vesicle) 釋出,上述現象與修格蘭氏症候群患者血清中含有大量的抗 Ro/SSA 抗體與抗 La/SSB 抗體相符合2,6。
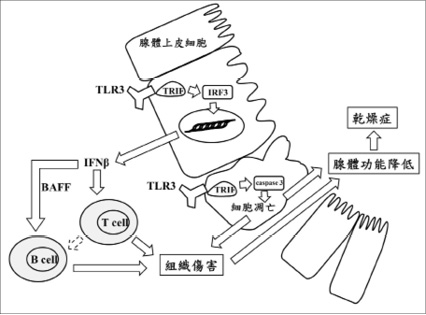
圖一 修格蘭氏症候群免疫反應路徑簡圖 (繪圖;郭廷濠)
三、其他因素
(一)病毒感染
雖然爭議尚存,但病毒感染可能與修格蘭氏症候群的誘發有關,如:C型肝炎病毒 (hepatitis C virus)、第一型人類嗜T淋巴球病毒 (T lymphotropic virus type I)、克沙奇病毒 (coxsackieviruses) 及艾伯斯坦-巴爾病毒或稱 EB 病毒 (Epstein-Barr virus,EBV) ,其中又以 EB 病毒最被廣泛討論,因為當 EB 病毒感染時,會刺激人體的先天免疫系統,並藉由B淋巴細胞活化因子活化B細胞3,此現象可以與上述患者上皮細胞大量表現 TLR3受體 (TLR3受體可調節先天免疫),藉干擾素 β、B 淋巴細胞活化因子活化B細胞造成組織傷害相呼應2。
(二)吸菸
除了病毒感染外,吸菸亦被認為是修格蘭氏症候群及其他自體免疫疾病 (紅斑性狼瘡與肌炎) 的危險因子2。
參、修格蘭氏症候群的診斷與臨床表現
一、診斷
2002年美國-歐洲共識小組 (American-European Consensus Group,AECG) 修訂的準則持續使用到最近,時至今日由於對修格蘭氏症候群的致病機轉逐漸明瞭,更為明確的標的使生物製劑得以發揮效用,所以需要更有效的方式執行臨床試驗,因此於2016年美國風濕病協會 (American College of Rheumatology,ACR) 和歐洲抗風濕病協會 (European League Against Rheumatism,EULAR) 共同提出新的 ACR/EULAR 分類準則7。
ACR/EULAR 分類準則2016年版有五個評估項目,各項目有相對應之分數,當患者的總分 ≧ 4即認定其為修格蘭氏症候群的患者,其中,該準則與2002年版最大之相異處為提高偵測出陽性抗 Ro/SSA 抗體與於組織切片有病灶區之淋巴球唾液腺炎 (sialadenitis) 且 focus score ≧ 1 focus/ 4 mm2這兩項目的比重 (3分)(表一)7。其他改變方面,如:排除條件去除已有淋巴瘤的病人、排除條件新增 IgG4相關疾病、將眼睛染色評分提高至5分、認為其他抗體 (包括 La/SSB) 特異性不高而不使用,僅採用抗 Ro/SSA 抗體等7,8。
表一 ACR/EULAR 分類準則2016年版7

二、臨床表現
(一)乾燥症狀 (sicca symptoms)
口腔及眼睛乾澀為修格蘭氏症候群患者的典型症狀2。口腔乾燥會造成患者需要配流質才能吞嚥較乾的食物、味覺改變、口腔燃燒感、無法像以前依樣流利說話、半夜常需起來喝水等2,另外,口腔乾燥易造成蛀牙,因此患者須注意口腔衛生避免牙周疾病的發生1。乾性角膜結膜炎 (keratoconjunctivitis sicca) 為修格蘭氏症候群患者眼睛的主要症狀,乾性角膜結膜炎會造成角膜及連結上皮的傷害,進一步加速淚膜 (tear-film) 破壞與淚液狀態的改變1,造成病人常感覺眼部癢或有砂礫感,患者可能會有無法佩戴隱形眼鏡或需使用人工淚液等情況2。
(二)一般症狀、全身症狀與併發症
修格蘭氏症候群患者的一般症狀有發燒、體重減輕及疲倦 (fatigue)2,其中以疲倦最常發生且被認為與干擾素有關5。
全身症狀與併發症則非常多元,有皮膚、關節、肺部、心血管、腎臟泌尿、神經、造血系統等,其中皮膚相關症狀有皮膚血管炎 (cutaneous vasculitis)、紫斑症 (purpura) 等;關節相關症狀以滑膜炎 (synovitis) 或關節炎 (arthritis) 最為常見;腎臟方面則有間質性腎炎 (Interstitial nephritis) 及膜性增生性腎絲球腎炎 (membranoproliferative glomerulonephritis)2。
(三)淋巴瘤 (lymphoma)
修格蘭氏症候群為一淋巴球浸潤疾病,特別是B淋巴細胞,因此約5-10%的患者最終會轉變成粘膜相關淋巴組織淋巴瘤 (mucosal-associated lymphoid tissue lymphoma,MALT lymphoma)5,唾腺、腮腺 (parotid gland) 為修格蘭氏症候群患者最常發生 MALT 淋巴瘤之區域,但其他區域 (如胃部、肺部、眼睛) 亦可能發生淋巴瘤2。
肆、修格蘭氏症候群的治療
以下就症狀治療藥物及全身性治療藥物做分述:
一、症狀治療藥物
症狀治療以改善口腔及眼睛乾燥為主1,2。
(一) 抗膽鹼類藥物 (muscarinic agonists)
抗膽鹼類藥物或稱分泌促進劑 (secretagogues),如:pilocarpine 或 cevimeline,係以抑制蕈毒鹼受體為作用方式,刺激修格蘭氏症候群患者的唾腺及淚腺分泌2。中重度口腔乾燥,但仍具有腺體功能的患者,以 pilocarpine 口服5 mg 每6小時給予或以 cevimeline 口服30 mg 每8小時給予,可以達到療效與副作用間的平衡,另外 pilocarpine 或 cevimeline 亦具有改善眼睛乾燥的效果,本類藥物的副作用可能有流汗、潮紅、頻尿、頭痛、噁心、鼻炎、咽炎、腹瀉及腹部疼痛1。
(二)抗發炎藥物
本類藥物以 ciclosporin 為主,具有抗發炎功效,使用於中重度修格蘭氏症候群之乾性角膜結膜炎 (keratoconjunctivitis sicca) 患者,以一天兩次0.05%或0.1%局部給予 ciclosporin,使用時間不超過6個月1,2。
(三)其他
唾液具有潤滑、緩衝及抗菌功效,因此唾液分泌不足會影響口腔健康狀況,在輕微唾液分泌不足的患者,可使用人工唾液 (saliva substitutes)、潤滑劑 (lubricating agents) 及無糖口香糖 (sugar-free gum) 來增加唾液與刺激唾腺分泌1。
淚液方面,需先排除其他可能造成眼睛乾燥的藥物,以人工淚液每日兩次以上為眼睛乾燥的第一線治療,建議使用含有玻尿酸 (hyaluronate) 或羧甲基纖維素 (carboxymethylcellulose) 且無防腐劑成分之人工淚液1。另外,潤滑軟膏 (lubricating ointments) 作用時間長,建議使用於睡前可改善患者視力受損1。
二、全身性治療藥物
可再分為非生物製劑及生物製劑類做細述:
(一)非生物製劑類
雖然缺乏強力的證據,但非生物製劑,如 prednisone 及 hydroxychloroquine,可能仍使用於修格蘭氏症候群患者的治療2。
類固醇藥物 prednisone 的使用係基於其具有改善其他自體免疫疾病 (如關節炎與皮膚疾病) 之效果,本藥物長期使用需考量骨質疏鬆、糖尿病、體重增重及血脂異常等副作用2;hydroxychloroquine 亦可用於關節痛、關節炎與皮膚疾病7,其具有抑制前述之 TLR 訊息傳遞路徑而來,可藉抑制 TLR 訊息路徑而減少干擾素等促發炎 (pro-inflammatory) 細胞激素的生成2。
(二)生物製劑類
1. Rituximab
Rituximab 為抗 CD-20 B 淋巴球受體之單株抗體,被核可用於治療B細胞淋巴瘤,具抑制 CD-20 B 淋巴球受體,利用抗體或補體依賴細胞毒性造成細胞凋亡7,rituximab 靜脈注射給予1 g 於修格蘭氏症候群患者可以改善疲倦、唾腺與淚腺之功能1,但目前 rituximab 的使用仍為藥品仿單標示外使用 (off-label use)2。
2.其他
Belimumab 為一 BAFF 之單株抗體,使用於全身性紅斑性狼瘡,以10 mg/kg 的劑量使用在修格蘭氏症候群患者的臨床試驗中展現出療效7。Ocrelizumab、obinutuzumab 與 ofatumumab 為其他抗 CD-20 B 淋巴球受體之單株抗體,目前已被使用於B細胞淋巴瘤的患者7。Epratuzumab 則為抗 CD-22 B 淋巴球受體單株抗體,在試驗中顯示可改善修格蘭氏症候群患者的疲倦症狀2。
伍、結語
修格蘭氏症候群為一自體免疫疾病,係由於淋巴球浸潤而破壞腺體造成患者的乾燥症狀,其主要病生理機轉理論 (自體免疫上皮炎) 的提出,使修格蘭氏症候群的致病機轉更加明瞭及標的更加明確,更多的生物製劑得以使用於治療修格蘭氏症候群。另外,也因為臨床試驗需求,ACR/EULAR 分類準則2016年版被提出,提高了某些評估項目的比重,使更多潛在患者可以被納入試驗並接受治療,將有更多患者得以受惠。
Sjögren's Syndrome
Ting-Hao Kuo
Meiga Pharmacy
Abstract
Sjögren's syndrome(SS) is a progressive autoimmune-related disease. Destruction of gland function by lymphocytes infiltration result in sicca symptoms. To date, the most support theory is ‘autoimmune epithelitis’. That is, epithelial cells act as atypical antigen-presenting cell and constantly express TLR3 let themself become central regulators. The 2016 ACR/ EULAR classification criteria are applied because of more understanding about the pathophysiology of SS, and needing for conducting clinical trials. Not only current treatment (pilocarpine and cevimeline), but novel biological therapies will be available in the future.
參考資料:
1.Saraux A, Pers JO, Devauchelle-Pensec V: Treatment of primary Sjögren syndrome.Nat Rev Rheumatol. 2016;12(8):456-71.
2. Brito-Zerón P, Baldini C, Bootsma H et al: Sjögren syndrome. Nat Rev Dis Primers. 2016;2:16047.
3. Nocturne G, Mariette X: B cells in the pathogenesis of primary Sjögren syndrome.Nat Rev Rheumatol. 2018;14(3):133-145.
4. Nocturne G, Mariette X: Advances in understanding the pathogenesis of primary Sjögren's syndrome. Nat Rev Rheumatol. 2013;9(9):544-56.
5. G Obermoser, V Pascual: The interferon-α signature of systemic lupus erythematosus.Lupus. 2010; 19(9): 1012-1019.
6. Okuma A, Hoshino K, Ohba T et al: Enhanced apoptosis by disruption of the STAT3-IκB-ζ signaling pathway in epithelial cells induces Sjögren's syndrome-like autoimmune disease.Immunity. 2013;38(3):450-60.
7. Nicoletta Del Papa, Claudio Vitali: Management of primary Sjögren's syndrome: recent developments and new classification criteria.Ther Adv Musculoskelet Dis. 2018; 10(2): 39-54.
8. Franco Franceschini, Ilaria Cavazzana, Laura Andreoli et al: The 2016 classification criteria for primary Sjogren's syndrome: what's new? BMC Med. 2017; 15: 69.
通訊作者:郭廷濠/通訊地址:台北市文山區保儀路23號
服務單位:美迦藥局藥師/聯絡電話:(O) 02-29362815

