摘要
肺動脈高血壓是一種漸進式且致命性的疾病而且其預後普遍較差,升高的肺動脈壓、肺部血管阻力以及肺部血管重新塑型可能會導致右心室肥大甚至右心衰竭,而這些因素都有可能增加病患的危險性及死亡率。儘管目前肺動脈高血壓確切的機轉還有待更進一步之釐清,但是肺部中許多內生性物質之間的失衡已被確定。傳統對抗肺動脈高血壓的藥物已有抗凝血劑、鈣離子阻斷劑、利尿劑、強心劑,但仍需發展更多的治療藥物及其複合療法,因為此一疾病的死亡率依然相當的高。近年實證醫學相關研究指出,內皮素接受體拮抗劑、磷酸二酯酶第五型抑制劑以及環前列腺素可用於治療肺動脈高血壓,另一方面,許多相關系列的實驗及研究亦指出血清素系統和Rho激酶可能是未來發展肺動脈高血壓藥物新的方向。
關鍵字: 肺動脈高血壓、內皮素接受體拮抗劑、磷酸二酯酶第五型抑制劑、PAH、ERA、PED5-I
壹、前言
肺動脈高血壓(PAH)是種罕見疾病及漸進式的疾病,一般會出現的病徵包含:休息時肺動脈壓持續性維持在>25 mmHg或是運動時肺動脈壓持續性維持在>30 mmHg,此外它會增加肺部血管阻力、組織結構增生進而影響肺部循環,而導致右心室肥大甚至衰竭造成死亡。肺動脈高血壓的詳細發病機制目前並無相當明確的定見,但近來許多學者的研究中亦發現於PAH的情況下,肺部血管內的血管收縮物質/增生性的物質(如:endothelin)和血管舒張性物質/抗增生性物質(如:prostacyclin and nitric oxide)以及其它會影響血液凝集的因子會產生失衡的現象,如圖一,而造成血管收縮、結構改變、增生等反應。
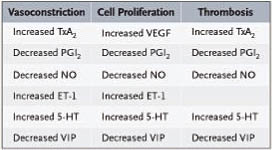
圖一 影響血管收縮、舒張及血液凝集之因子
儘管學者多年來已投入大量研究,但對於PAH仍是無法完全治癒,目前認可的治療藥物包含了prostanoids,endothelin受體結抗劑和磷酸二酯酶第五型抑制劑,以及鈣離子阻斷劑和NO,這些藥物有改善症狀,運動能力,血流動力學的效果。上述血管舒張劑中,像是prostacyclin、鈣離子通道阻斷劑和endothelin-1受器拮抗劑使用在肺動脈高血壓,但效果都有限,其原因不外乎於缺乏肺部血管選擇性,其中,鈣離子阻斷劑易造成持久性的全身性低血壓、暈厥及心血管衰竭須注意。
在PAH的病患中,吸入性NO可以產生選擇性的肺部血管舒張作用而且並不影響系統性血壓,然而NO在空氣中不安定,會自行氧化成NO2,故在使用期間要連續監測NO和NO2濃度,相當不方便,且長期高濃度的使用會增加變性紅血球(methemoglobinemia)產生的機會;若合併使用高濃度氧氣會合成二氧化氮,對肺組織有強烈的刺激作用,令哮喘、咳嗽及喘鳴惡化。此外,用於PAH的藥物尚有抑制 PDE5(phosphodiesterase type5)的藥物如sildenafil,可使肺動脈中cGMP的濃度上升而紓緩肺部壓力, 其它較新的治療方向,如Rho-kinase抑制劑 fasudil 也有文獻指出,用於對於吸入性NO以及對nifedipine反應差的PAH病患可明顯降低其肺部血管阻力而且不會造成系統性低血壓的情況1,其次,抑制SER/SERT(Serotonin /Serotonin transporter)系統的相關研究亦日益增多,但仍需更多證據來驗證其確切的療效及副作用,本文簡要回顧目前肺動脈高血壓的治療方式,以為臨床用藥之參考。
貳、成因及危險因子
有關PAH確切且詳細的致病原因目前尚無定論,但許多研究都指出此一惡性的心肺疾病與肺部血管的重塑(remodeling)、收縮、血小板功能異常與血栓生成有關,目前有學者更進一步提出,家族性PAH患者與其本身bone morphogenetic protein receptor II(BMPR2)基因產生變異有關,儘管其他更精確的致病原因並不明確,不過會導致PAH的危險因子或可能的誘發因素已被確認,像是使用包括fenfluramine及dexfenfluramine在內的減肥藥物三個月以上即會增加產生PAH的危險性23倍以上,甚至有文獻更進一步的指出,使用這些減肥藥物減重的風險遠大於不使用而繼續肥胖的人2,而其它危險因子有肝硬化、具有PAH家族病史、受愛滋病毒感染和使用cocaine等。
參、肺動脈高血壓之預後
一般來說此疾病的預後非常的差,通常嚴重的患者存活率< 3年,2002年有學者統計出PAH患者在產生心臟突然停止情況後存活率是很低的,更令人遺憾的是,於法國近期診斷出肺動脈高血壓的674名患者中只有88%一年後還存活,再者,2007年一篇系統性回顧的文章中提及,整合13個研究含1,999名PAH患者參予的隨機試驗中顯示,沒有被治療(給予安慰劑)的868名受試者中,有10%的病人其病情於12-18週的試驗過程中產生臨床上的惡化,2.3%的受試者死亡,再者,給予藥物治療的1,131名受試者產生臨床上的惡化者為4.8%,有1.4%的受試者死亡,令人遺憾的是針對死亡的數據並無統計學上的差異(level 2 evidence)3,不過就減低PAH患者死亡的危險而言2009年Chest發表的一篇文章中指出,選擇性的血清素再回收抑制劑似乎可改善PAH患者死亡的危險4,當然,這些相關的證據都顯示目前人類對於PAH的挑戰還有相當大的進步空間。
肆、 肺動脈高血壓之實證藥物治療現況
PAH可能由原有肺血管疾病、慢性肺疾、心臟疾病、肝臟疾病所引發;也可能在胸腔或心臟手術後發生,因此在過去對於PAH患者使用的藥物包含了O2、鈣離子阻斷劑、抗凝血劑、利尿劑、強心劑等等,這些藥物的搭配使用,可用來減輕患者的心肺負擔,此外吸入性NO與IV heparin搭配使用也曾成功的應用於新生兒的原發性PAH,但其安定性及耐受性需要注意,然而隨著新的藥物不斷開發、演進,現今的用藥方向也較以往有了一些不同,增加了許多新的選擇以及搭配,以下列出目前具有實證結果的用藥。
一、 Endothelin receptor antagonists(ERA):bosentan、sitaxsentan、ambrisentan
ERAs可改善PAH病人的呼吸困難和運動能力(level 1 evidence),一般用於NYHA Class II – IV者,(WHO/NYHA心肺功能等級請參閱表一及表二)在一篇針對ERAs治療PAH的系統性文獻回顧顯示(具有11項隨機試驗),參予的1457名PAH患者使用ERAs明顯改善了患者的運動能力、WHO/NYHA功能性評估和一些心肺血液動力學指數(肺動脈壓、肺部血管阻力、cardiac index)。結果亦指出選擇性ERA可降低死亡率,上述隨機試驗除了探討ERAs對PAH的效果外,亦有bosentan(非選擇性ERAs)與sildenafil治療PAH的比較,但此二者在治療效果上並沒有顯著差異,至於肝毒性方面則是各給藥組都沒有顯著性的不同5。此外,於另一篇為期18週針對選擇性ERA:sitaxsentan用於PAH的試驗中,每天100 mg sitaxsentan可改善6分鐘行走距離及減少肺功能惡化(level 1 evidence),受試的247名PAH病患(12-78歲),分別給予安慰劑、sitaxsentan 50 mg/天、sitaxsentan 100 mg/天、bosentan(前四週一天二劑62.5 mg,之後以一日二次125 mg投予),評估其6分鐘行走距離,結果顯示出服用bosentan及sitaxsentan 100 mg/天的病患,其增加的行走距離明顯多於安慰劑組,於試驗最後(第十八週)評估受試者之肺功能,發現服用安慰劑與sitaxsentan 50 mg者,有13%惡化、服用sitaxsentan 100 mg有2%(NNT 9)惡化,服用bosentan者有9%惡化6。其它FDA核准的PAH用藥如ambrisentan於實證上亦能改善PAH患者之運動能力。(level 1 evidence)。
表一 WHO之肺動脈高血壓患者功能分級
分級 |
定義 |
I |
運動不受限制,一般運動不會造成呼吸困難或倦怠、胸痛及暈厥 |
II |
運動輕微受限,休息時舒服。一般運動會造成呼吸困難或倦怠、胸痛及暈厥 |
III |
運動明顯受限,休息時舒服。比日常活動量稍少即會造成呼吸困難或倦怠、胸痛及暈厥 |
IV |
進行任何活動時均有症狀,休息時可能出現呼吸困難或倦怠,且任何活動都會使身體不適更嚴重,並且出現右心衰竭徵候。 |
表二 NYHA之肺動脈高血壓患者功能分級
分類 |
定義 |
I |
日常生活不會造成症狀 |
II |
休息時沒有症狀,但日常活動有症狀 |
III |
休息時沒有症狀,但輕微活動就有症狀 |
IV |
休息時有症狀發生 |
二、Prostacyclins(一般用於NYHA Class III – IV)
IV prostacyclin 可改善運動能力、心肺功能(New York Heart Assocation functional class)和心肺血液動力學長達12週;根據IV prostacyclin或類似物的隨機試驗的系統性文獻回顧發現1175名原發性PAH在試驗期間(3天-52週)可改善運動能力,其中4項試驗中亦呈現出可改善心肺循環及心肺功能;口服prostacyclin的試驗中顯示可改善運動能力3-6週以上,但也有服藥12個月時,其運動能力及其他結果並無明顯差異之案例;其它像是皮下給予treprostinil或吸入給予prostacyclin都可改善運動能力、心肺循環。
(一)、Epoprostenol sodium(Flolan)
Epoprostenol sodium(Flolan)持續性IV給予用於治療原發性PAH可改善存活率;162名原發性PAH患者在epoprostenol 治療平均36.3個月後,於一年、二年、三年的存活率分別為87.8%、76.3%、62.8%,高於過去資料的27.4-30%7,epoprostenol一般是透過輸注幫補經由中央靜脈導管輸注給予,起始劑量為2 ng/kg/min,可依需求慢慢調整劑量,部分患者需要使用到200 ng/kg/min的高劑量,常見副作用有噁心、嘔吐、頭痛、腹部疼痛、低血壓…等,treprostinil (Remodulin)為合成的prostacyclin,近年FDA核准其用於皮下持續注射以對抗PAH及NYHA class II-IV的症狀。
(二)、Aerosolized prostacyclin(Iloprost)
吸入Iloprost對於嚴重PAH可緩解其症狀,在臨床上的隨機試驗中,有16.8%的患者使用Iloprost10 mcg/mL每天6-9次後,其六分鐘步行測試增加至少10%,而且針對使用後的患者再次以NYHA功能性評估,評估結果亦有改善,而且沒有惡化或死亡8。
三、 Phosphodiesterase type-5 inhibitor PDEV-inhibitor)
(一)、Sildenafil(Viagra)
在一系列針對sildenafil的臨床試驗中發現,sildenafil可能具有改善PAH患者的肺功能(level 2 evidence),另一方面,在合併治療方面,於IV epoprostenol的患者中加入口服sildenafil治療後也發現可能會因此增加患者的運動能力以及減少病狀的惡化(level 2 evidence)9, 其它PDEV-inhibitor像是tadalafil(Cialis)在實證上也有相類似的結果,它被發現可以改善患者的運動能力、延遲臨床症狀惡化、改善生活品質(level 2 evidence)
伍、結論
儘管在許多研究當中顯示出這些治療效果能改善存活率並降低住院率、運動能力及改善血液動力學,但目前的治療策略,仍是不夠的,因為死亡率仍然很高,肺功能和血流動力學障礙仍然廣泛出現在許多病人當中,這些因素都一再的顯示出抗PAH的治療方式還具有很大的進步空間,除了搭配實證醫學上的許多證據找出更有效的用藥組合,來提升患者治療的效益及醫病品質,更須著手於新藥開發,目前研究中較為新穎的PAH治療目標是針對Rho Kinase(ROCK)以及血清素系統(SER/SERT)加以抑制,來達到抗PAH的目的,許多基礎醫學研究一再地證明,無論是ROCK或是SER/SERT都在PAH的病理學上扮演重要的角色,而這些相關藥物的研究相信也會對未來對抗PAH帶來新的契機!
參考資料:
1. Fukumoto Y, Matoba T, Ito A, et al: Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart (British Cardiac Society) 2005; 91(3): 391-2.
2. Abenhaim L, Moride Y, Brenot F, et al: Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 1996; 335(9): 609-16.
3. Helman DL, Jr., Brown AW, Jackson JL, Shorr AF. Analyzing the short-term effect of placebo therapy in pulmonary arterial hypertension: potential implications for the design of future clinical trials. Chest 2007; 132(3): 764-72.
4. Shah SJ, Gomberg-Maitland M, Thenappan T, Rich S. Selective serotonin reuptake inhibitors and the incidence and outcome of pulmonary hypertension. Chest 2009; 136(3): 694-700.
5. Liu C, Chen J, Gao Y, Deng B, Liu K. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane database of systematic reviews (Online) 2009(3): CD004434.
6. Barst RJ, Langleben D, Badesch D, et al: Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006; 47(10): 2049-56.
7. Kapp C. WHO chief announces surprise move to stand down. Lancet 2002; 360(9334): 695.
8. Olschewski H, Simonneau G, Galie N, et al: Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347(5): 322-9.
9. Galie N, Ghofrani HA, Torbicki A, et al: Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353(20): 2148-57.
Currrent Therapy of Pulmonary Hypertension
Chien-Heng Huang, Chin-Kuo Huang, Chia-Ching Shih, Wei Lee, Mau-Fu Wang Department of pharmacy, Taoyuan Armed Forces General Hospital at Taoyuan County, Taiwan
Abstract
Pulmonary arterial hypertension(PAH)is a progressive and fatal disease with poor prognosis. PAH, characterized by an progressive elevation of pulmonary arterial pressure, vascular resistance and vascular remodeling which may cause right ventricular hypertrophy, heart failure, and increases mortality of patients. However, the precise mechanism of PAH still remains to be elucidated, but an imbalance between vasoconstrictor/proliferative agents(e.g. endothelin)and vasodilator/antiproliferative substances(e.g. prostacyclin and nitric oxide)have been identified in the lung vasculature. Although anticoagulant agents, calcium channel blocker, diuretics, and inotropic agent are currently used for treatment of PAH, more effective treatment or combination therapy needs to be developed. Recent Evidence-based medicine suggests that endothelin receptor antagonists(ERA), phosphodiesterase type V inhibitor(PDE5-I) and prostacyclin may be promising drugs in the treatment of pulmonary arterial hypertension.
In addition, series of experimental and clinical studies suggested that serotonin system and Rho kinase is an attractive target for the development of therapeutics to treat PAH.

