摘要
高雪氏症 (Gaucher disease) 是一種溶小體儲積症,主要是因為基因缺陷造成體內代謝酵素異常,導致醣脂質在體內累積而引起病變。酵素替代療法 (Enzyme replacement therapy, ERT) 與基質減量療法 (Substrate reduction therapy, SRT) 是目前最主要的兩種藥物治療方式,但兩者在使用上都還有許多限制,這也是未來藥物發展需要克服的部分。
關鍵字: 高雪氏症、酵素替代療法、Imiglucerase、肝脾腫大、神經病變
壹、前言
高雪氏症是一種與體內代謝相關的罕見疾病,是在1882年由法國醫生 Philippe Gaucher 從一位32歲的女性屍體上發現,他觀察到病人有肝脾腫大的情形,因而認定這是脾臟癌表現的一種形式,但直到1965年,才由其他學者確立高雪氏症的致病機轉及臨床症狀表現,相關的治療方式才得以開始發展、研究1。
貳、分類
高雪氏症屬於最常見的溶小體儲積症 (Lysosomal Storage Diseases, LSD),因為其致病機轉與醣脂質在巨噬細胞中的溶小體 (lysosome) 累積有關2。LSD 大多是基因異常而導致酵素的活性降低,使得體內某些化學物質異常的堆積,進而導致全身器官如內臟、神經、骨骼、皮膚等部位發生病變1。
參、流行病學
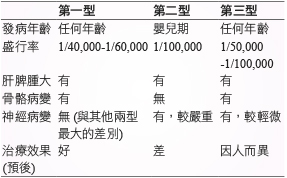
全世界的盛行率大約是每40,000人中會有一個人是高雪氏症的患者,但會因疾病類型與種族的不同而有所差異。依照病人臨床症狀的不同,高雪氏症可區分為三種類型。第一型:是目前最常見的一種,95%為西方國家的病人,尤其是艾希肯納茲猶太族裔 (Ashkenazi-Jewish),大約每450人就有一個人會是高雪氏症的患者,每十人就有一人帶有異常的突變基因3;第二型:為最罕見的一種,約每十萬人中才會有一人是此類型;第三型:常發生在中東、印度、中國與太平洋沿岸的國家,未來有機會取代第一型,成為全球最常見的高雪氏症類型2。而台灣目前因為病例數較少,所以盛行率不明4。
肆、病理學
高雪氏症屬於一種隱性體染色體的遺傳疾病,其致病機轉是由於調控體內 Glucocerebrosidase (葡萄糖腦苷脂酶,又可稱作 Glucosylceramidase 或 Acid beta-glucosidase,簡稱 GBA) 的基因發生突變,導致酵素活性降低,無法將體內的葡萄糖腦苷脂還有與神經病變有關的 glucosylsphingosine 代謝掉 (圖一),這些過多的醣脂質就會在巨噬細胞裡的溶小體累積,透過骨髓的切片檢查可以觀察到巨噬細胞呈現兩種可供疾病鑑別的特徵,一個是細胞核會集中在細胞的某一側;另一個則是因為醣脂質的累積,會使得細胞質的部分看起來就像一張被揉皺的紙,這樣的細胞型態稱為高雪氏細胞 (Gaucher cell),高雪氏細胞會在身體各個部位累積而引起該部位的病變。常會產生病變的器官包括:肝臟、脾臟、骨骼、骨髓、大腦、淋巴結等等1。
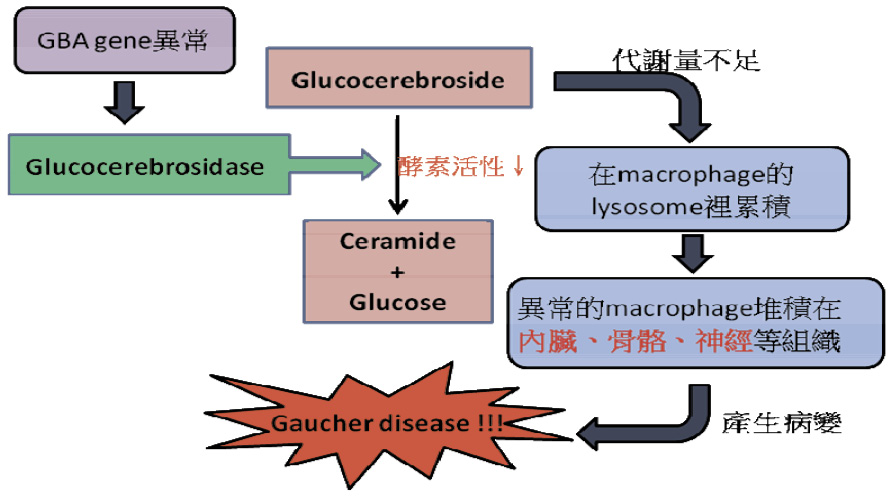
圖一 高雪氏症的致病機轉簡圖1,3
基因突變的部分主要是發生在第一條染色體的 q21區段異常1,根據目前的統計有超過200種 GBA 基因的突變與高雪氏症有關,其中超過80%是發生在單一核苷酸的取代 (single-nucleotide substitutions)1,從 International Collaborative Gaucher Group (ICGG) 所做的統計結果來看,常見的突變位置有 c.1226A > G allele (N370S mutation)、c.1448T > C allele (L444P mutation)、c.84dupG allele (84GG mutation) 這三種。在統計中發現佔53%的 N370S mutation 與非猶太裔歐洲人、艾希肯納茲猶太族裔常發生的第一型非神經病變型較有相關性,L444P mutation 則是在北歐國家常發生的第二、三型神經病變有關3。
伍、臨床表現及診斷
高雪氏症主要的症狀有肝脾異常腫大、貧血、血小板低下、骨質疏鬆與神經病變等等,以神經病變區分第一型與二三兩型間的判斷標準,通常病人在神經病變的初期會先出現眼球運動上的問題,像是斜視、核上性凝視麻痺 (supranuclear gaze palsy),後來還有可能出現運動功能失調、癡呆、心智遲緩、肌陣攣性發作 (myoclonus) 等嚴重症狀1,3,7。以下就三種類型的差異分別說明 (表一)。
一、第一型
唯一不會出現神經病變的類型,主要的症狀有肝脾腫大、貧血、骨骼疏鬆,這類病人通常對於藥物治療 (酵素補充療法) 的效果良好,病人通常都能活得比較久並且維持不錯的生活品質1,4。
二、第二型
又稱作急性神經病變型 (acute neuronopathic type),一般病人在出生3-6個月內就會發病,其神經病變的症狀較為嚴重且發展迅速,病人常在2歲之前就已經過世,導致無法觀察到骨骼病變的產生。此類型藥物治療的效果並不好,目前只能採取支持性療法來緩和症狀1,4。
三、第三型
屬於慢性神經病變型 (chronic neuronopathic type),神經病變的症狀較晚發生且較輕微,又依照臨床表徵的不同再分成3a、3b、3c 三種亞型,Type 3a 主要為神經病變的相關症狀,但肝脾腫大的情況較不嚴重;Type 3b 在內臟、骨頭的症狀比較嚴重,神經病變較少且進展緩慢。在某些族群 (例如位於瑞典北部的 Norrbottnian) 有特別高的盛行率;Type 3c 則是與心血管的病變較有關聯。這類病人在治療上常因神經病變發展程度而造成治療效果上的差異,較早出現神經病變的病人症狀都會較嚴重,治療的預後也較差1。
表一 高雪氏症三種類型的比較1,4
在診斷上,最主要的方式就是測定周邊白血球的 glucocerebrosidase 活性是否下降1,通常高雪氏症患者體內的酵素活性會小於正常值的30%5,也可透過檢驗數據、影像學檢查、理學檢查觀察病人有沒有出現高雪氏症常見的症狀,比較特殊的檢查則是將在高雪氏症病人體內會異常增高的化學物質作為生物指標,例如:幾丁三糖酶 (chitotriosidase)、CCL18、ACE、TRAP (tartrate resistant acid phosphatase,抗酒石酸酸性磷酸酶) 等,其中 chitotriosidase 是最優先考慮的檢測項目,但因為有6-8%的病人體內 chitotriosidase 酵素活性也有缺陷,因此就會換成 CCL18、ACE、TRAP 作為評估標準,其中 CCL18是一種在高雪氏症患者中會異常增加的趨化因子 (Chemokine),主要與體內的免疫調節有關6,7。此外,基因突變位置的分析也可以協助預測疾病的臨床表現,以及突變基因帶原者 (carrier) 的檢測1。
陸、藥物治療
隨著對高雪氏症的致病機轉越來越清楚,除了透過骨髓移植、脾臟切除等方式來緩解疾病的惡化,還有其他治療的方式供病人選擇。高雪氏症的藥物治療目標就是要減少醣脂質 (如:glucocerebroside、glucosylsphingosine) 在病人體內的堆積,目前以酵素替代療法 (ERT) 與基質減量療法 (Substrate reduction therapy, SRT) 為主要治療方式。以下從目前常用的藥物治療方式及未來發展方向進行介紹。
一、 酵素替代療法 (Enzyme replacement therapy, ERT)
酵素替代療法就是藉由將結構修飾後的酵素打入病人體內,使得體內醣脂質的代謝增加,而達到治療效果。但因為無法通過血腦障壁,僅限於用在第一型高雪氏症1。
Alglucerase (Ceredase, Genzyme Corporation) 是第一個被研發出來的治療藥物,於1991年獲得 FDA 許可上市,是經由人類胎盤進行萃取純化,在胎盤取得不易、萃取步驟繁雜的情況下,使藥物產量受到限制;在1994年透過重組 DNA 技術製造出來的 Imiglucerase (Cerezyme, Genzyme Corporation),Imiglucerase 是一個具有497個胺基酸的葡萄糖腦苷脂酶類似物,其與 Alglucerase 結構上不同是在第495位置上的胺基酸以 histidine 取代 arginine,兩者同樣都在寡醣鏈 (oligosaccharide chains) 加上甘露醣來修飾結構,使得巨噬細胞上的受體 (甘露醣受體、甘露醣六磷酸受體) 能夠辨識藥物並與之結合,然後透過胞吞 (endocytic pathway) 的方式進入到溶小體,最後促進醣脂質的代謝8,9。
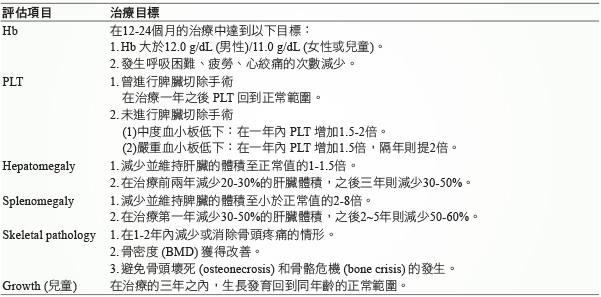
Imiglucerase 的治療劑量,是依據病人的症狀嚴重度選擇起始劑量,劑量範圍從2.5 U/kg 每週施打三次,到60 U/kg 每一到四周施打一次都有治療效果。在一個12位病人的雙盲平行試驗 (parallel trial) 中,兩組分別使用15 U/kg 每兩周一次、2.5 U/kg 每周三次在貧血、血小板低下的改善是無顯著差異的;在相同累積劑量,但分為每兩周及每四周施打一次的分析中,在貧血、肝脾腫大的治療上也無顯著差異5。目前在文獻中大多建議以60 U/kg 每兩週施打一次的方式來治療,再依照病人的症狀改善程度、副作用或過敏反應產生來進行劑量調整9。Imiglucerase 的治療目標請見 (表二)。
表二 Imiglucerase 用於第一型高雪氏症病人的治療目標5
在副作用方面,兩者皆有注射部位不適、噁心、嘔吐、腹部不適產生,尤其在使用 Alglucerase (14%)、Imiglucerase (16%) 的第一年內會產生 IgG 的抗體,抗體會影響到藥物的藥動學性質,對於效果並不會有太大的影響,需要注意的是體內有 IgG 抗體反應的病人,可能在後續藥物治療的過程中發生類似過敏的休克反應,所以在治療第一年內需定期監測病人體內抗體的產生,若出現抗體反應,在後續的治療上要特別注意出現過敏情形9,10。
近幾年有 Velaglucerase alfa (VPRIV,2010年獲得 FDA 許可)、Taliglucerase (ELELYSO,2012年獲得 FDA 許可) 被研發出來,但目前在台灣只有 Imiglucerase 通過衛福部許可2,4。
二、 基質減量療法 (Substrate reduction therapy, SRT)
是經由抑制製造葡萄糖腦苷脂的合成酶 glucosylceramide synthase,減少醣脂質合成的方式來治療,ERT 是促進醣脂質代謝,兩種治療的目的都是為了減少體內醣脂質的累積 (圖二),目前只用在第一型高雪氏症對 ERT 耐受性不佳 (容易出現注射部位不適、嚴重過敏反應)的病人,作為 ERT 的替代療法。因為這類藥物的相關研究還不夠完整,SRT 只能用於成年人,孕婦、兒童、年長者、嚴重肝腎功能異常的病人都不建議使用 SRT2,12。
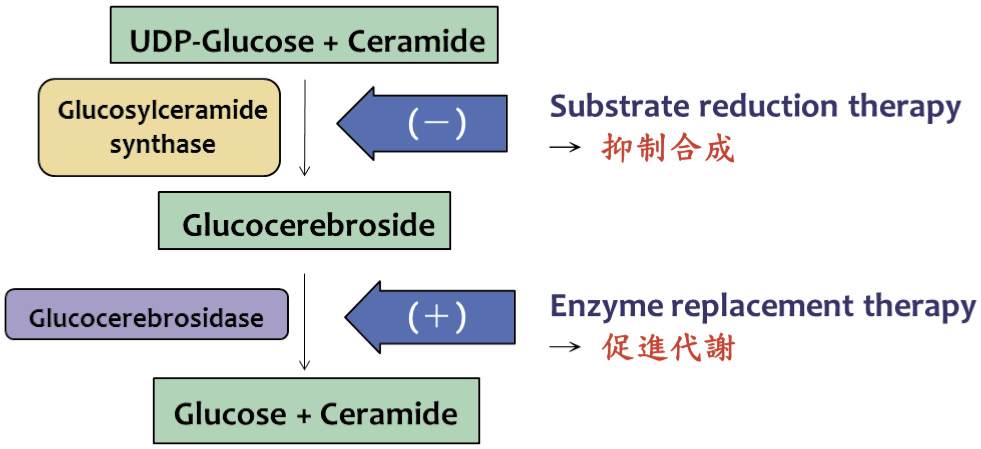
圖二 ERT 與 SRT 治療機轉比較8
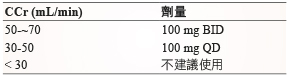
藥物有 Miglustat (Zavesca,2003年獲得 FDA 許可)、Eliglustat (Cerdelga,2014年獲得 FDA 許可),兩種皆為口服藥物,在使用劑量上,Miglustat 的劑量範圍從100 mg BID/QD-200 mg TID,最常使用劑量為100 mg TID,可依病人的情況和副作用進行調整。Miglustat 因不經由肝臟代謝,主要是以原型藥的形式透過腎臟排出體外,腎功能不全的病人就必須依照肌酐酸清除率 (CCr) 來調整劑量 (表三)。Eliglustat 則是依據病人體內 CYP2D6的代謝效率來給予不同劑量,廣泛與中間代謝型 (extensive and intermediate metabolizers) 族群使用劑量為84 mg BID;弱代謝型 (poor metabolizers) 為84 mg QD12。在一個 phase III 的隨機試驗中,對一個已接受過 ERT 治療的成年病人,Eliglustat 可以維持其在貧血、器官腫大上的治療效果5。
表三 Miglustat 在腎功能不全病人的劑量調整12
此類藥物會引起腹瀉、噁心、腹痛等腸胃道副作用,其中 Miglustat 因為會抑制腸道酵素,所以特別容易引起胃腸副作用11,如腹瀉 (85-100%)、脹氣 (29-50%)、腹痛 (18-67%)、便秘 (8%) 等。導致病人服藥順從性差。其他還包括頭痛、抽筋、異常的體重減輕,需定期進行神經學檢查避免周邊神經病變12。
三、支持性療法 (Supportive care)
針對臨床症狀進行治療,例如:骨質疏鬆可使用雙磷酸鹽藥物、維生素D;嚴重貧血或血小板低下 (thrombocytopenia) 則可經由輸血治療,過去常會將病人脾臟做部分或完全的切除,以減少脾臟對於紅血球、血小板的破壞,但因為切除脾臟後常會造成免疫功能下滑,導致因感染引發敗血症,現況若因為脾臟嚴重腫大導致貧血,且對酵素替代療法的效果不彰,才會考慮將病人的脾臟進行切除1。
四、 造血幹細胞移植 (Hematopoietic stem cell transplantation, HSCT)
骨髓移植原是高雪氏症病人常使用的治療方式1,但因移植需考量配對的問題,且手術後常有感染或排斥反應的風險,因此不是一種很理想的治療方式。隨著科技的發展,可以透過臍帶血進行幹細胞移植,來減少排斥反應與骨髓配對上的問題8。
柒、未來發展
未來須持續研發治療方法,像是基因療法、酵素增強療法 (Enzyme enhancement therapy, EET) 等方式,針對現有的酵素替代療法藥物無法通過血腦障壁來治療神經病變,或許可以透過增加藥物溶解度,或利用載體來攜帶藥物通過血腦障壁,來提升藥物在腦部的濃度,達到治療效果,也是目前相關研究努力的目標之一1。
Treatment of Gaucher Disease
Song-Yuan Chung1, Pei-Shan Cheng2, Shu-Zhen Yang2, Pei-Ling Tsai2, Lin-Chu Liao2
School of Pharmacy of China Medical University1
Department of Pharmacy, National Taiwan University Hospital Yun-Lin Branch2
Abstract
Gaucher disease is a kind of Lysosomal Storage Diseases (LSD). Because the deficiency of metabolic enzymes caused by genetic defects, leading to accumulation of glycolipid in the body caused by lesions. Enzyme replacement therapy (ERT) and Substrate reduction therapy (SRT) are the most important two kinds of drug therapy, but both have the limitation of use. Therefore, it’s a part of future development to overcome.
參考資料:
1.Aabha Nagral: Gaucher Disease, Journal of Clinical and Experimental Hepatology. 2014; 4(1): 37-50.Gaucher disease: Pathogenesis, clinical manifestations, and diagnosis, UpToDate
2. National Gaucher Foundation http://www.gaucherdisease.org/
3. Derralynn Hughes, MD. Gaucher disease: Pathogenesis, clinical manifestations, and diagnosis. In: UpToDate, Post TW(Ed), UpToDate, Waltham, MA. (Accessed on November 17, 2016.)
4. 李榮明、李炳鈺、謝右文:高雪氏症臨床治療之展望。衛生福利部,2006。
5. Christine Serratrice, Sebastian Carballo, Jacques Serratrice, et al: Imiglucerase in the management of Gaucher disease type 1: an evidence-based review of its place in therapy, Dove Press journal. 2016; 11: 37-47
6. Rolf G. Boot, Marri Verhoek, Maaike de Fost, et al: Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention, Blood. 2004; 103(1): 33-9.
7. Derralynn Hughes, MD. Gaucher disease: Initial assessment, monitoring, and clinical course. In: UpToDate, Post TW(Ed), UpToDate, Waltham, MA. (Accessed on November 17, 2016.)
8. Giancarlo Parenti, Claudio Pignata, Pietro Vajro, et al: New strategies for the treatment of lysosomal storage diseases (Review), International Journal of Molecular Medicine. 2013; 31: 11-20.
9. Drug result page. Imiglucerase. Truven health analytics MICROMEDEX® Solutions, 2016. Available at Accessed 8 December, 2016.
10. Derralynn Hughes, MD. Gaucher disease Treatment. In: UpToDate, Post TW(Ed), UpToDate, Waltham, MA. (Accessed on November 17, 2016.)
11. Balwani M, Burrow TA, Charrow J, et al: Recommendations for the use of eliglustat in the treatment of adults with Gaucher disease type 1 in the United States, Molecular Genetics and Metabolism. 2016 Feb;117(2):95-103.
12. Drug result page. Miglustat. Truven health analytics MICROMEDEX® Solutions, 2016. Available at Accessed 8 December, 2016.
通訊作者:鄭佩珊/通訊地址:雲林縣斗六市雲林路二段579號
服務單位:台大醫院雲林分院藥劑部藥師/聯絡電話:(O) 05-5323911 ext 5186

