摘要
亨丁頓氏舞蹈症是一具種族特異性的遲發體染色體顯性遺傳之神經退化疾病,其基因缺陷係位於第4條染色體短臂16的位置,因 CAG 三聯核酸重複序列異常擴增造成其 HTT 蛋白具有過長的多麩醯胺酸鏈,繼而影響正常的細胞生理機轉,造成大量的細胞功能異常。患者可能展現出運動、認知、精神疾病方面的症狀,診斷上主要藉由基因檢測,係檢測 HTT 基因是否有 CAG 序列異常擴增為主。目前尚無足以改善亨丁頓氏舞蹈症進程的治療,因此在症狀治療上更顯重要,在過去治療上多仰賴 tetrabenazine,而目前新核可的藥物 deutetrabenazine 在許多方面都優於原藥物,因此開啟治療亨丁頓氏舞蹈症的新序章。
關鍵字: 亨丁頓氏舞蹈症、CAG 三聯核酸重複序列、HTT 蛋白、tetrabenazine、deutetrabenazine
壹、前言
優雅的舞蹈令人如癡如醉,但失序凌亂的舞姿,卻是某些疾病患者噩夢的序曲。亨丁頓氏舞蹈症 (Huntington's chorea) 或稱亨丁頓氏症 (Huntington Disease;HD),於1872被醫師 George Huntington 發現而後得其名,特徵為患者會有非自發性的舞蹈現象,是一遲發之體染色體顯性遺傳的神經退化疾病1。
其盛行率具種族特異性,於歐洲人約為0.01%,亞洲人約為0.001%,非洲人則相對更少,其特色為發病年齡多於中年 (約35-45歲),但少數患者仍有可能於20歲前發病 (約10%),表現出少年型 (juvenile form) 亨丁頓氏舞蹈症1,2。
藉由連鎖分析 (linkage analysis) 得知,亨丁頓氏舞蹈症之基因缺陷係位於第4條染色體短臂16的位置 (4p16.3),該位又特稱為 Huntingtin (HTT) 基因,因 CAG 三聯核酸重複序列 (CAG triplet repeat) 異常擴增造成其 HTT 蛋白具有過長的多麩醯胺酸鏈 (polyglutamine;poly Q),變異之 HTT 蛋白的累積,繼而影響正常的細胞生理機轉1-3。
貳、 亨丁頓氏舞蹈症的病生理學致病機轉
HTT 基因經過轉錄及 RNA 加工後,會產生兩種 mRNA,一種為能轉譯出完整長度(full-length) 之 HTT 蛋白的 mRNA,另一為僅具有外顯子1之短片段 mRNA (HTT exon1 mRNA),該短片段 mRNA 能轉譯出 HTT 外顯子1蛋白 (HTT exon1 protein),具完整長度之 HTT 蛋白亦可能藉由蛋白水解切割 (proteolytic cleavage) 轉變成 HTT 外顯子1蛋白2。
HTT 基因中以外顯子1最為重要,且具不正常長度之 CAG 三聯核酸重複序列亦位於該外顯子1內,僅由 HTT 基因中之外顯子1所轉譯出的 HTT 外顯子1蛋白細部組成可分為三區,第一區為蛋白質N端具有17個胺基酸之區域 (17個胺基酸分別為 MATLEKLMKAFESLKSF),稱為 HTTNT或 N17,第二區為 CAG 三聯核酸重複序列轉譯出的 poly Q,第三區為富含脯氨酸之區域 (proline-rich domain;PRD),此 HTT 外顯子1蛋白被認為是導致亨丁頓氏舞蹈症主要蛋白2,4。
形成 HTT 外顯子1蛋白被認為是亨丁頓氏舞蹈症早期的關鍵步驟,HTT 外顯子1蛋白可以進入細胞核內,在核內形成聚集的現象干擾正常細胞的轉錄功能;HTT 外顯子1蛋白亦可在細胞質聚集破壞蛋白動態平衡 (proteostasis),影響蛋白動態平衡會造成突觸功能異常 (synaptic dysfunction)、粒線體毒性 (mitochondrial toxicity) 及能量失衡、軸突傳導 (axonal transport) 障礙等,且 HTT 外顯子1蛋白的聚集現象在全腦區皆有可能發生,並隨著 poly Q 的增加,HTT 外顯子1蛋白越容易聚集,造成大量的細胞功能異常 (圖一)2。
另外,HTT 外顯子1蛋白的異常聚集,使亨丁頓氏舞蹈症與其他神經退化疾病有相關的連結,例如阿茲海默症等2。HTT 外顯子1蛋白所造成的突觸功能異常被認為主要為干擾在紋狀體中的突觸後多巴胺神經功能以及丘腦皮質 (thalamocortical) 的麩胺酸神經傳遞系統,干擾多巴胺神經功能造成多巴胺受體過度活化,當多巴胺受體過度活化時就會產生不自主的舞蹈症狀,而影響麩胺酸神經傳遞系統,則造成丘腦皮質有過多的麩胺酸傳遞物質,形成運動過度 (hyperkinetic) 的相關症狀5。
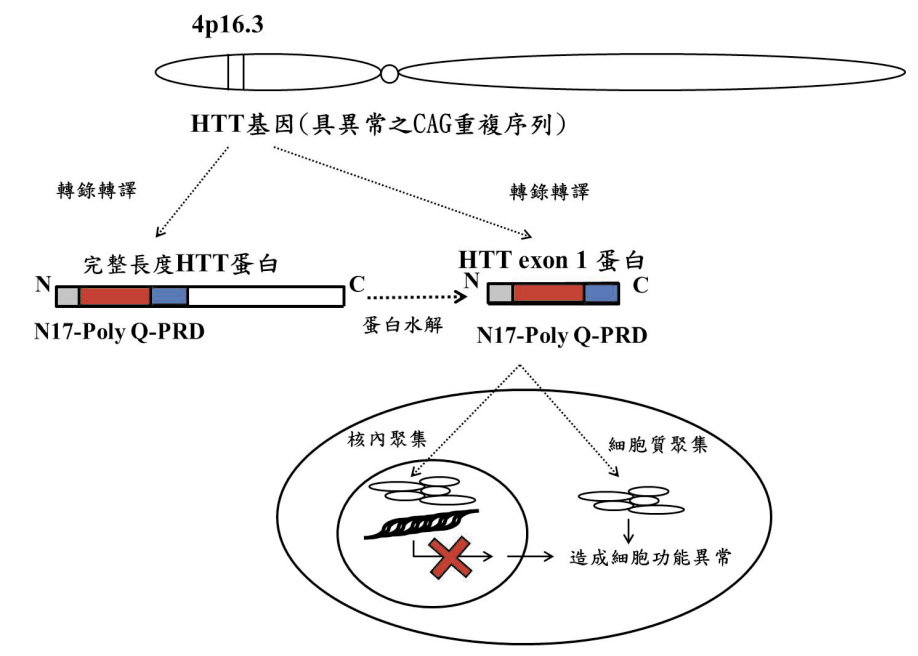
圖一 亨丁頓氏舞蹈症的致病機轉 (繪圖:郭廷濠)
參、亨丁頓氏舞蹈症的診斷與臨床表現
臨床上診斷亨丁頓氏舞蹈症主要是藉由:
一、臨床評估
主要可藉由觀察具家族史之患者的不隨意運動 (involuntary movement) 與緩發性之情緒障礙而來,徵兆為持續惡化的運動、心理及認知異常1,或是藉亨丁頓氏舞蹈症評定量表 (Unified Huntington's Disease Rating Scale;UHDRS) 對運動功能 (motor function)、認知功能 (cognitive function)、異常行為 (behavioral abnormalities)、功能效能 (functional capacity) 四大面向進行評分,來評估其臨床表現6,7。
患者在運動症狀可能展現舞蹈症、運動過度症狀、肌張力不全症、運動功能減退、運動緩慢、僵硬、顫抖、喪失姿勢反射等,認知症狀方面有衝動、認知扭曲、缺乏注意力或易分心、學習能力障礙、組織能力障礙、溝通障礙等,精神疾病症狀則有睡眠障礙、憂鬱、焦慮、煩躁、冷漠、強迫症、性功能過度、精神病、失智症等1,7。
二、家族史
家族史有助於亨丁頓氏舞蹈症之診斷,因亨丁頓氏舞蹈症患者的子代有一半的機會會遺傳有此疾病之基因,但由於亨丁頓氏舞蹈症為遲發性疾病,病發時患者多半已有其子代,因此家族史非理想之手段,臨床上診斷亨丁頓氏舞蹈症之最主要手段為基因檢測1。
三、基因檢測
基因檢測係檢測 HTT 基因是否有 CAG 序列異常擴增的情況,平均而言,歐洲人序列約為18.4至18.7重複、亞洲人約為17.5至17.7重複、非洲人為16.9至17.4重複,非洲人序列重複較少也對應其盛行率較其他兩人種為低2。序列重複數在27至35之間時,具序列異常擴增之個體可能或可能不進展為亨丁頓氏舞蹈症,因此,此長度之序列重複又稱為不定的 (indeterminate) 或中間的 (intermediate) 序列,當序列重複數大於等於36時,則發病的可能性大大提高,尤其是序列重複數大於等於40的個體2,3。
序列重複數在40至59間之個體為典型亨丁頓氏舞蹈症的患者,而當序列重複數在60至95間,則個體會表現出少年型亨丁頓氏舞蹈症1。
四、神經影像與其他檢測
亨丁頓氏舞蹈症患者的病理檢查主要可見逐漸退化的基底核 (basal ganglia),且其他腦區如黑質 (substantia nigra)、大腦皮質 (cerebral cortex)、海馬迴 (hippocampus) 等亦有退化的情況1。
肆、亨丁頓氏舞蹈症的治療
目前尚無有效的治療足以改善亨丁頓氏舞蹈症的進程,因此,除了給予病人適當的衛教外,症狀治療亦十分重要,用於改善患者的運動功能、增進其生活品質 (quality of life) 及保護患者的安全。
一、 tetrabenazine (TBZ) 與 deutetrabenazine
最常被使用在亨丁頓氏舞蹈症的藥物有 TBZ 與抗精神病藥物,其中,TBZ 是被美國食品藥物管理局 (FDA) 核准治療亨丁頓氏舞蹈症的藥物,其機轉為可逆性抑制第二型囊泡單胺類轉運體 (vesicular monoamine transporter type 2;VMAT2) 之中樞單胺消耗劑,且對於多巴胺的選擇性高於正腎上腺素,TBZ 的初始劑量由每日12.5 mg 開始,一周後可將劑量提高,最高至極量為100 mg 或患者無法承受其副作用,而其副作用有靜坐不能、憂鬱、頭暈、帕金森氏症等,此外,任何服用 TBZ 的病人須注意其憂鬱和自殺傾向7,8。另外,TBZ 的劑量在高於50 mg 以上時亦須考慮患者之代謝酶 CYP2D6 基因型的差異 (快速代謝或慢速代謝)7,8。
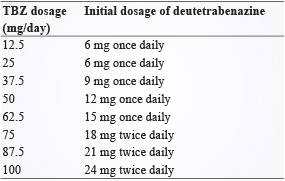
於2017年,TBZ 的氘取代類似物 (deuterated analog) deutetrabenazine 亦被 FDA 核准使用於舞蹈症治療,成為第二個用於亨丁頓氏舞蹈症的許可藥物,其作用機轉與 TBZ 相同,僅在關鍵位置上,將氫原子取代為其同位素氘,致使 deutetrabenazine 在藥物動力學上的表現能較 TBZ 佳,deutetrabenazine 在治療的劑量也能夠較 TBZ 低,其起始劑量為每日6 mg,一周後可將劑量提高,最高劑量為每日48 mg (當劑量大於每日12 mg 時建議以一天兩次給予),因此提高了 deutetrabenazine 的安全性,另外,其他較 TBZ 優勢方面為 deutetrabenazine 有緩釋 (slow-release) 劑型、較 TBZ 少的副作用及對亨丁頓氏舞蹈症同等或更好的控制效果7,9。當病人需要在 TBZ 與 deutetrabenazine 的治療上做轉換時,可以參考轉換時 deutetrabenazine 的起始劑量轉換表 (表一),此外,如同 TBZ,使用 deutetrabenazine 的患者一樣須注意其憂鬱和自殺傾向,以及其亦須考慮代謝酶 CYP2D6的基因型差異與藥物交互作用 (會干擾 CYP2D6之藥物)9。
表一 TBZ 劑量對應 deutetrabenazine 起始劑量轉換表9
二、 多巴胺受體拮抗劑 (dopamine antagonists)
本類藥物主要為抗精神病藥物 (neuroleptics),是最常使用於改善亨丁頓氏舞蹈症患者情緒障礙的藥物,可進一步分為典型 (typical) 與非典型 (atypical),在典型藥物可以使用如:haloperidol,非典型藥物則有:olanzapine、risperidone、quetiapine 等,其中以 olanzapine 與 risperidone 較常被使用,另外,新一代的非典型藥物 aripiprazole 在部分小型研究中展現出與 TBZ 相當程度的治療效果7,8。
抗精神病藥物的副作用亦可能造成亨丁頓氏舞蹈症患者的部分運動症狀,如厭食、遲發性肌張力不全症與眼動危象等,但此現象可以藉由降低劑量或是停用藥物來改善7。
三、苯二氮平類 (benzodiazepines)
本類藥物主要在於緩解患者的焦慮與舞蹈症的狀態,如 clonazepam 以每日5.5 mg 給予被認為可以抑制舞蹈症狀,另外,本藥物的副作用為鎮靜7,8。
四、 麩胺酸受體拮抗劑 (glutamate antagonists)
本類藥物有 NMDA 受體抑制劑的 amantadine 以及干擾紋狀體之麩胺酸釋放與降低神經毒性的 riluzole,amantadine 係以每日400 mg 的劑量給予,當使用到更高劑量時,需考量對症狀緩解是否有益處,riluzole 則是以每日200 mg 給予8。
伍、結語
亨丁頓氏舞蹈症目前尚無完全治癒的可能,其多半以症狀治療為主,希望提升病人的生活品質與保護患者的安全,過去使用的 TBZ、多巴胺受體拮抗劑、benzodiazepines 以及麩胺酸受體拮抗劑似乎已不再能滿足現今的治療,隨著近年研究對亨丁頓氏舞蹈症中的病生理機轉逐漸明瞭,以及 HTT 外顯子1蛋白的功能及其異常聚集等瞭解,使得許多新藥物能進一步研究用在治療亨丁頓氏舞蹈症,而目前新核可的藥物 deutetrabenazine,機轉與 TBZ 相同,但在藥物動力學、劑量、安全性等許多方面都優於 TBZ,也使亨丁頓氏舞蹈症的治療開啟了新的序章。
Huntington’s Chorea
Ting-Hao Kuo
Meiga Pharmacy
Abstract
Huntington’s chorea(HD) is a race-specific, autosomal-dominant, late-onset neurodegenerative disease. The mutation for HD is on chromosome 4p16.3 with the over-expanded CAG triplet repeat, which encodes the Huntingtin proteins with abnormal polyglutamine segment and leads to cellular function impairment. The features of HD are motor function, cognitive function and psychotic disorder. DNA diagnostic testing for expanded CAG repeat is mean analysis in patients with HD. To date, there still are not promising candidates for curing HD, therefore symptoms management becomes more important in treating HD. Before 2017, tetrabenazine (TBZ) was the only approved drug for HD treatment by FDA. Nowadays, deutetrabenazine is the second drug approved for treating HD, which not only is considered to be better then TBZ, but may disclose novel therapeutic strategies for HD.
參考資料:
1.Yapijakis C: Huntington Disease: Genetics, Prevention, and Therapy Approaches.Adv Exp Med Biol.2017; 987: 55-65.
2. Bates GP, Dorsey R, Gusella JF, et al: Huntington disease.Nat Rev Dis Primers. 2015;1:15005.
3. Pagan F, Torres-Yaghi Y, Altshuler M: The diagnosis and natural history of Huntington disease. Handb Clin Neurol. 2017; 144:63-67.
4. Aiken CT, Steffan JS, Guerrero CM, et al: Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity.J Biol Chem. 2009; 284(43): 29427-36.
5. Coppen EM, Roos RA:Current Pharmacological Approaches to Reduce Chorea in Huntington's Disease.Drugs. 2017; 77(1):29-46. doi: 10.1007/s40265-016-0670-4.
6. Huntington Study Group: Unified Huntington's Disease Rating Scale: Reliability and Consistency.Mov Disord. 1996; 11(2): 136-42.
7. McCusker EA, Loy CT: Medical management of motor manifestations of Huntington disease.Handb Clin Neurol. 2017; 144: 141-150.
8. Samuel Frank: Treatment of Huntington's Disease. Neurotherapeutics. 2014; 11(1): 153-160.
9. Heo YA, Scott LJ: Deutetrabenazine: A Review in Chorea Associated with Huntington's Disease.Drugs. 2017;10.1007/s40265-017-0831-0.
通訊作者:郭廷濠/通訊地址:台北市文山區保儀路23號
服務單位:美迦藥局藥師/聯絡電話:(O) 02-29362815

