摘要
藥師在執行整個臨床試驗中扮演很重要的角色。本文將以醫院研究用藥專責管理藥師的觀點,以某醫學中心為例,探討在臨床試驗中藥師應不斷更新的知能及受試者保護觀念。希望藉由經驗分享 (包含從臨床試驗執行前的準備工作到進行時的注意事項),提升台灣醫院藥師對臨床試驗的概念,以確保受試者的權利、安全與福祉。
關鍵字: 臨床試驗、臨床試驗藥品管理專責藥師、美國臨床研究受試者保護評鑑協會(AAHRPP)、受試者保護計畫
壹、前言
藥品臨床試驗的目的在於探討新藥在人體的有效性和安全性。臨床試驗通常可區分為四個分期階段:第一階段為測試人體對新藥的忍受度,第二階段則針對療效與安全性測試,第三階段擴大試體數目,以統計證實新藥的有效性,第四階段目的在於對副作用的長期追蹤1。藥師的角色在執行整個臨床試驗的前、中、後期,常見職務包含了人體試驗委員會委員、臨床試驗審議委員會、數據與安全監測委員會、臨床試驗藥品管理專責藥師等2。本文將以醫院研究用藥專責管理藥師為主,以某醫學中心為例探討在臨床試驗中藥師應具備不斷更新的研究知能、國際化視野與受試者保護觀念,以及如何落實於藥局作業,以期能提升受試者安全與福祉。
我國衛生福利部食品藥物管理署 (簡稱食藥署) 於2018年6月7日在國際醫藥法規協和會 (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use,ICH) 神戶會議成為藥政法規單位正式會3,此重大成果代表我國醫藥界為符合國際法規標準的多年努力與貢獻受到國際認可。ICH 於1990年成立起,積極制定新藥研發可共同接受的品質、療效、安全、跨領域等76項規範。二十多年來 ICH 規範已成為先進國家新藥研發的黃金標準4,所有致力於新藥開發及參與跨國臨床試驗的先進國家無不依此規範訂定臨床試驗執行的相關準則。事實上,在臨床試驗的發展中,台灣是亞洲最早參考美國食品藥物管理局 (FDA) 精神建立符合 ICH 法規環境的國家5。衛生署 (為衛生福利部前身) 於九十四年一月六日訂定「藥品優良臨床試驗準則」(Good Clinical Practice, GCP)6即是參考八十五年公告之「藥品臨床試驗規範」與國際上通用的國際醫藥法規協合會之 ICH E6 (Guidance for Industry E6 Good Clinical Practice: Consolidated Guidance) 所制定的7。
二十多年來,在政府所建立之國際醫療水準的法規基石上,台灣的醫療水準與臨床試驗的發展也逐步提升,在國際上享有良好的信譽與可信度,使我國無論是在早期臨床試驗或多國性臨床試驗的參與案件數明顯增加。由於醫學中心家數多,病人樣本數充足,加上研究能量品質優良,2015年台灣參與跨國臨床試驗的排名躍居全球第25名8。
近十年來包含韓國、中國在內的亞洲各國皆策略性的全力發展臨床試驗產業。為了能夠在競爭愈趨激烈的全球臨床試驗市場中保有競爭優勢,所有執行臨床試驗的各家醫學中心無不積極提升臨床研究實力,從硬體設備、專業人才的培育、臨床試驗中心的成立到申請國際研究認證,這些努力在本質上都是為受試者的保護提供了充分的保障。
貳、研究用藥與受試者保護
美國臨床研究受試者保護評鑑協會 (Association for the Accreditation of Human Research Protection Programs,以下簡稱 AAHRPP) 為2001年於美國成立的非政府單位非營利組織。AAHRPP 評鑑是目前國際上對臨床研究最嚴格的評鑑標準。其認證的目的是確認機構內臨床研究受試者保護工作 (Human Research Protection Program;HRPP) 是否具有完善機制、充足資源以及建立良好的監管制度,足以讓受試者得到足夠的保護。
在醫院的體系中,臨床試驗藥局為 HRPP 組織單位之一 (圖一),肩負研究用藥管理與保護受試者用藥安全的責任。因此,如何建立研究用藥標準化管理方式,以提供專業的研究用藥服務,已然成為藥學專業的一門重要課題。
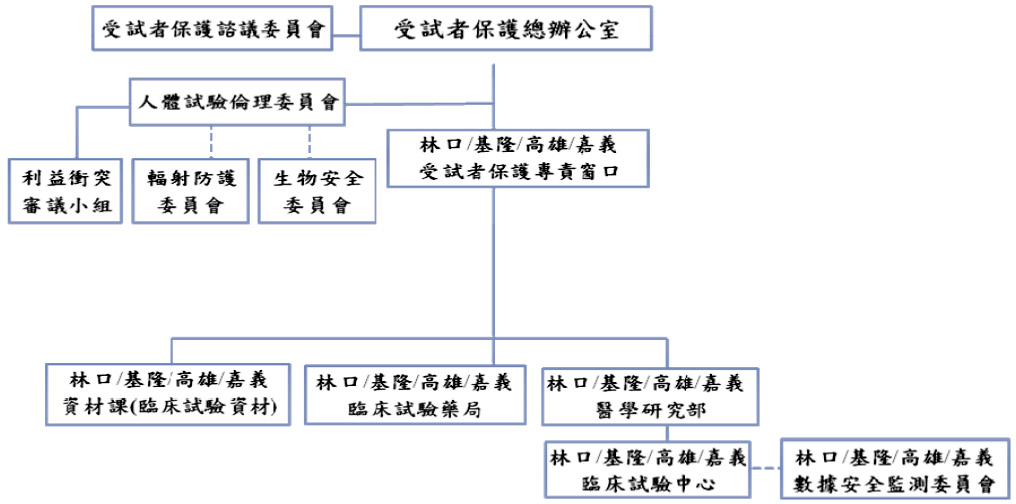
圖一 HRPP 組織架構
有鑑於此,美國醫院藥師學會 (American Society of Health-System Pharmacists, ASHP) 於2018年4月公布一份研究用藥管理指引 (ASHP Guidelines for the Management of Investigational Drug Products)9。指引的目的主要是讓臨床試驗藥局、新藥研發廠商等相關研究者,有一套管理研究用藥的標準化方法,期望機構正視專業臨床試驗藥局的存在與專責藥師設置的重要性。以下以某醫學中心為例,介紹臨床試驗藥局以標準化作業管理,強化藥師對臨床研究執行與管理的知能,進而提供更深入的研究用藥服務,以實踐對受試者保護的最終目的。
參、臨床試驗執行前的準備工作
一、制定規範化之作業規定
本院依據相關法規制定「人體研究暨受試者保護作業管理辦法」,以確保院內所有人員遵守相關法律及倫理標準,實踐人體研究計畫之審查、監督及受試者保護措施,並促使院內所有單位及研究者遵從相關規定,以保障參與研究的受試者之權益和福祉。專責藥師的業務規範則設立有「臨床試驗藥品管理作業準則」,內容涵蓋臨床試驗專責藥師之資格、職責、訓練並明定業務範圍:試驗藥品之管理、試驗藥品之調劑、試驗藥品之安全監測、試驗藥品之儲藏、試驗藥品之諮詢與用藥指導、其他試驗藥品相關之協調等事宜。此外更詳細規範臨床試驗藥品管理作業之共通性執行流程。
二、個別案件的專業訓練
各臨床試驗開始執行前,應先經由衛生主管機關與 IRB 核准,並為確保受試者的權益與研究者的相關義務,於進行試驗委託廠商與院方的合約書簽署後,再由計畫主持人或研究專員 (Clinical Research Associate,CRA) 與藥師約定計畫書內容及藥品編碼專業訓練會議,並須於約定日期之前依院方檢附資料清單準備相關文件給藥師審閱 (圖二)。藥師須事先確實審閱文件的正確性與完整性,並對計畫書內容進行了解,唯有會議前充分的準備工作,才能確保不遺漏任何重要的細節。
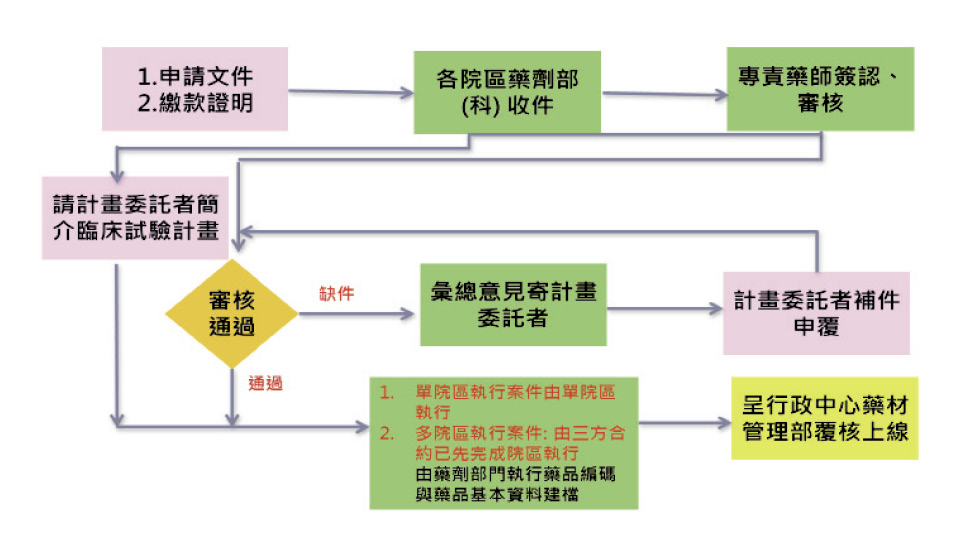
圖二 院內試驗藥品編碼收件及電腦上線審查流程
CRA 在訓練藥師的過程中,除了針對研究設計與藥品發放頻率、數量、劑量調整及發生不良反應的處理等加強說明之外,雙方對於藥品進藥流程、品質判定、藥品回收與表單填寫方式,皆須透過有效的溝通與協調過程來確立。藥師並依據討論結果,建立各案件的藥品管理作業流程,此流程將為日後藥師執行該案件的作業依據,以確保藥品記錄、儲存、調劑及運送等相關作業流程符合計畫書。
三、試驗藥品編碼與電子化審查流程
專業訓練完成後,藥師須進行試驗藥品的編碼設定,並通知該項試驗主持人,處方開立注意事項及配合事宜,以確保開立處方的正確性。藥品編碼的設定須以藥品外觀標籤所標示之品名、規格為依據,同時在雙盲試驗中則需要考慮研究設計的盲性維持,不可因為藥品名稱、開方數量的呈現而導致意外解盲。藥碼編定後的審查流程須經單位主管確認設定合理性後,同時連結批價、庫存等資訊系統後,始能開啟院區藥品檔進行試驗藥品管理與處方籤開立。醫院利用資訊化管理系統將繁瑣的藥品審核與建檔工作以最便捷的方式迅速建立,不但提高行政效率、增加收案競爭力,更縮短了受試者的用藥等待期。
肆、臨床試驗進行時的注意事項
一、藥品發放的審核機制
專責藥師發放試驗藥品之前,應先確認開方醫師是否為該案件主持人或協同主持人,而依據的標準為人體試驗倫理委員會 (Institutional review board, IRB) 之試驗同意核准函,若非函文上載明之參與醫師,不可開立該案試驗藥品。若有新增或刪除試驗醫師,藥師應將資訊更新於藥品管理作業流程,以做為給藥依據。此外,藥師應確認受試者是否已簽署受試者同意書,唯有已簽署同意書且符合資格之受試者才能領用試驗藥品。案例醫院使用資訊系統管控開立試驗藥品 (圖三),當主持人納入新受試者時,須上傳受試者同意書給 IRB,並由系統列印出受試者清單提供給藥師作為發藥依據,若受試者未簽署同意書,或主持人未將同意書上傳 IRB,系統將會管制不可開立試驗藥品。
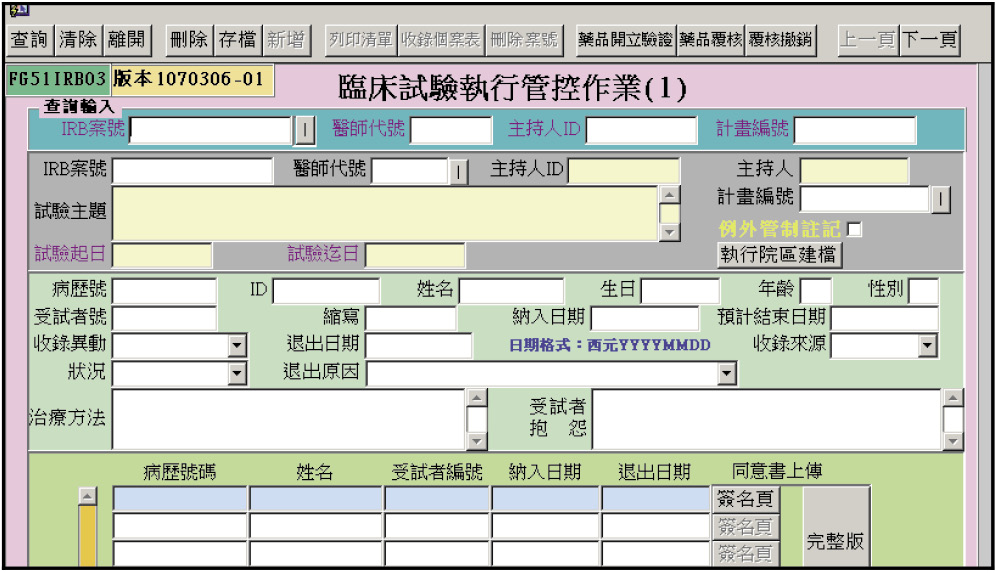
圖三 臨床試驗管控作業系統
試驗進行過程中,藥師除了審查用藥資格外,應對試驗計畫書及研究藥品充分了解,發放藥品時應能針對研究設計之領藥訪次 (Visit)、使用品項、劑量調整等確實審核,避免發生試驗不遵從事件。若在臨床試驗期間更新試驗計畫書及主持人手冊,藥師應確認變更案已經通過衛生主管機關與 IRB 核准,請 CRA 針對變更內容進行藥師訓練,並提供變更後文件及留存訓練記錄。藥師須將變更後的藥品管理及發放注意事項相關內容及藥品不良反應相關的安全性問題等,詳細記錄於藥品管理作業流程。
二、藥品管理與監控
醫院訂定之「人體研究暨受試者保護作業管理辦法」明訂試驗用藥應存放於臨床試驗藥品中心或藥局,由專責藥師管理。因此試驗藥品的點收、儲存、調劑、退藥回收等作業,皆須由專責藥師依據 GCP 與相關法規程序進行並留存書面文件。試驗藥品點收時,應注意藥品外觀標示之計畫書編號、藥盒號碼、數量、批號、效期等是否與到藥檢附文件內容吻合、藥盒是否有損毀或丟失及藥品運送期間溫度管控與判讀。如有異常情形,應先將該藥品上鎖隔離於具有適當溫度的空間,待與試驗廠商確認後才可放行使用或辦理退藥。藥品調劑時應確認處方簽內容之正確性與完整性,是否符合試驗計畫書內容之訪次、劑量及用法。交付藥品時應與研究助理再次核對受試者資訊、覆誦藥盒號碼或批號、確認藥品效期是否在受試者下次返診領藥之後。調劑表格填寫必須正確、完整、即時、字跡清晰。若須修改應以單線劃掉,但原記錄仍清晰可見,修改者於修改處簽上姓名及日期。當試驗中止或結束時,應請 CRA 到藥局與藥師實際對點藥品,確認藥品品項、數量後,簽署退藥相關文件,將藥品妥善包裝並連絡貨運公司進行回收。藥師應與貨運人員和核對計畫書編號,確認回收品項正確後,方可交付退藥,並注意留存退藥收執聯備查。
伍、結論
臨床試驗專責藥師應以法規為依據,以保護受試者為目的,進行試驗藥品管理相關作業。臨床試驗中藥師應具備不斷更新的研究知能與國際化視野,具備受試者保護觀念,以落實於藥局作業 (包含從臨床試驗執行前的準備工作到進行時的注意事項)。本文醫院積極追求臨床研究品質與保護研究受試者的權益,經過多時的努力,全體系人體試驗倫理委員會與各院區已同時通過 AAHRPP 認證,為全台首次7家醫院一起通過評鑑的機構。期望分享執行經驗,提升藥師能對臨床試驗的概念與增加受試者安全,進而建構符合自己醫院需求以受試者保護為目的研究用藥服務。
The Clinical Trial Management for The Purpose of Human Subject Protection
Hsuan-Lin Wen, Ching-Ling Tai, Chun-Lan Liu, Chiu-Ying Wu
Department of Pharmacy, Chang Gung Memorial Hospital, Kaohsiung
Abstract
Pharmacists have played a pivotal role in the management in the clinical research. In this paper, we was discussed the concept of knowledge and human subject protection in a medical center clinical research pharmacist opinion. We hope that through the experience sharing, pharmacists can enhance the concepts about clinical trials including the rights, safety, and welfare of subjects.
參考資料:
1.李慶三:Clinical Trial - What Is It。藥學雜誌2009;25:12-21
2. 施雅分、廖淑貞、陳薏如、簡素玉:藥師在臨床試驗中扮演的角色與功能。藥學雜誌2011;27:72~75
3. 衛生福利部:國際醫藥合作重大突破-我國成為國際醫藥法規協和會(ICH)會員。Available from: Https://www.mohw.gov.tw/cp-16-41553-1.html
4. 台灣醫藥品法規協會:ICH最新進展及其影響"之省思。Available from: Http://tsrap.org.tw/law-info_article-1.php?id=78
5. 台灣醫藥品法規協會:台灣臨床試驗競爭力再評估與建言系列(1)-臨床試驗中心。Available from: Http://tsrap.org.tw/news_article-1.php?id=76
6. 藥品優良臨床試驗準則,2014。
7. 國際醫藥法規協和會。Available from: http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html#6-2。
8. 楊泮池:培育中高階人才跳脫代工。輝瑞杏苑2016;10:5~6。
9. Stephen C. Kay, Darlette G. Luke, Helen R. Tamer: ASHP Guidelines for the Management of Investigational Drug Products. Am J Health-Syst Pharm. 2018; 75:561-73.
通訊作者:吳秋瑩/通訊地址:高雄市鳥松區大埤路123號
服務單位:高雄長庚紀念醫院臨床藥學科藥師/聯絡電話:(O) 07-7317123 ext 8410

