摘要
馬凡氏症候群 (Marfan syndrome, MFS) 是一種結締組織異常疾病,其病因與位於15號染色體的肌纖維蛋白基因 (fibrillin-1) 異常有關,疾病的臨床表現及嚴重度有很大的差異性,常涉及心血管及肌肉骨骼異常並且涉及多個器官系統,特別是最危及生命的主動脈剝離與主動脈瘤破裂,治療需定期監測追蹤、服用藥物或手術,藥物治療的目標在減緩主動脈擴大引起的致命風險。
關鍵字: 馬凡氏症候群、主動脈剝離、Marfan syndrome、fibrillin-1 gene、β-blockers、ARB
壹、前言
MFS 其病名取自1896年法國小兒科醫師 Antoine Marfan 首次描述此疾病,直到1991年由 Dietz 發現與疾病相關的基因1,盛行率約為三千至五千分之一,國內並無相關統計,沒有種族或性別偏好,某些病人有高於常人的身高或手臂臂長,病人常有主動脈根部擴大的相關疾病,其中主動脈剝離與主動脈瘤破裂是造成死亡的主因,部分猝死案例與之有關,經由藥物的治療,可減緩併發症發生及改善預後。
貳、病因學
一、致病機轉
典型 MFS 病人被發現位於15號染色體肌纖維蛋白基因 (fibrillin-1, FBN1) 突變,該基因有65個外顯子 (exons),外顯子26-27和31-32中的突變與疾病早期發作和嚴重度有關2,此基因與形成結締組織中的彈性纖維有關,fibrillin 是微纖維 (microfibrils) 的主要結構,提供水晶體懸靭帶、彈力蛋白與其他結締組織支持,若 fibrillin 發生異常,將破壞結締組織完整性,例如造成眼睛水晶體懸韌帶薄弱而脫位、主動脈壁薄弱而有剝離的危險。組織學表現包括主動脈壁中膜和彈性薄層破壞,使血管平滑肌細胞和彈性纖維局部凋亡消失,隨後在其中沉積蛋白樣物質,這些變化使主動脈變厚不易擴張,更易發生主動脈剝離3。
二、診斷與評估工具
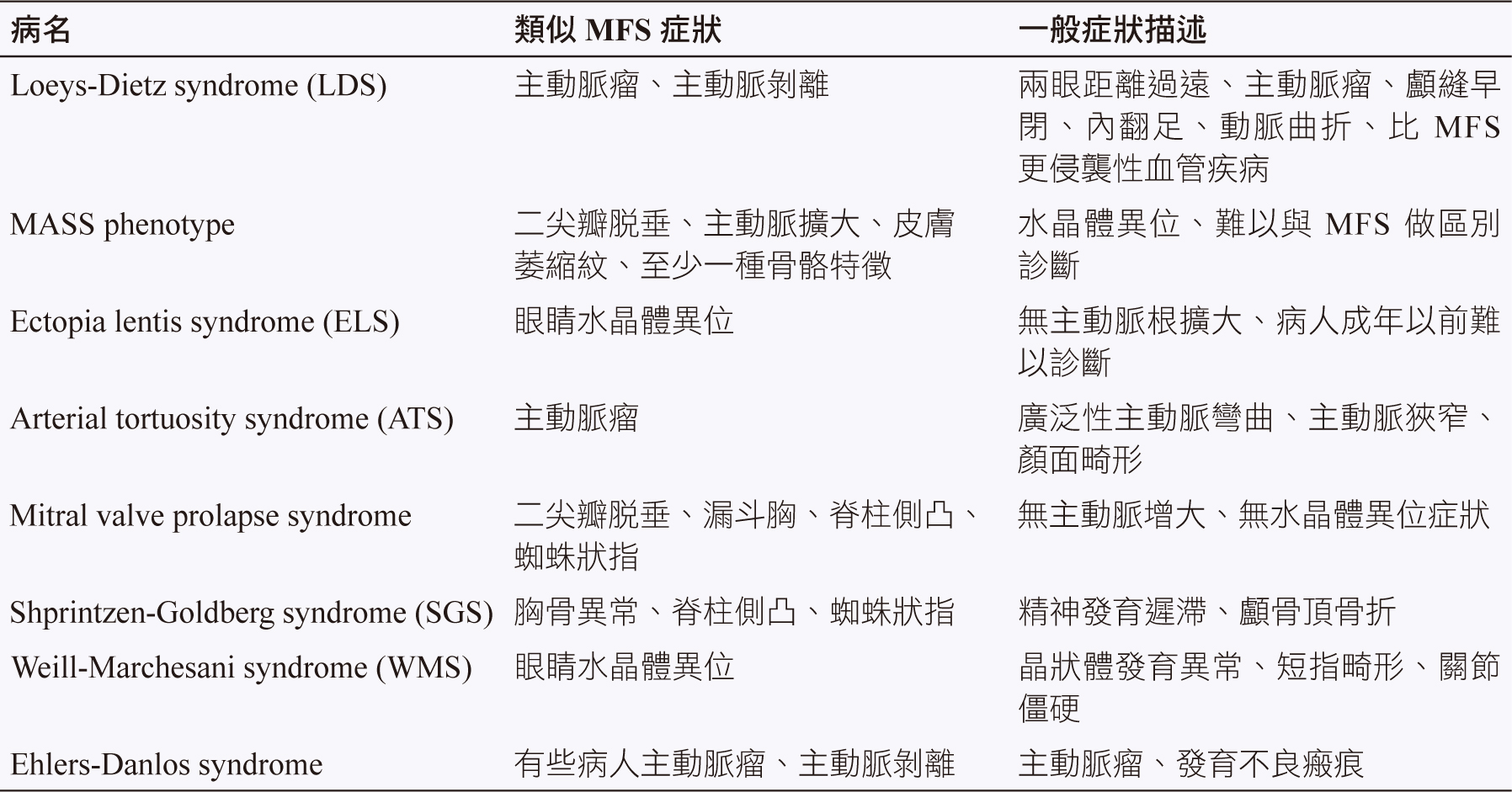
臨床表徵與許多其他遺傳疾病類似 (表一),所以不易做區別診斷,易造成過度診斷,依據2010年修正 Ghent 疾病分類準則 (表二及表三),把診斷重點放在主動脈根部擴大的程度、主動脈剝離及水晶體異位等症狀,另外包括檢測 FBN1 的突變4。分子遺傳學技術有助於診斷,但無法代替全面的追蹤評估,因為少數 (小於10%) 有典型 MFS 病人卻無 FBN1突變。
由於正常主動脈直徑大小是隨著年齡與體表面積而變化,對於主動脈根部評估標準5,是使用Z分數來表示,計算標準是假設病人體表面積來評估主動脈根部大小的線性關係 (Marfan基金會網站提供Z評分計算器)6。
參、臨床表徵
MFS 涉及人體多系統的疾病,在心血管系統方面,主動脈根部疾病包括動脈瘤擴大、主動脈回流及主動脈剝離,這也是造成 MFS 罹病率及致死率的主要原因,有50%主動脈擴大在病人兒童時期被發現,大約60%-80% MFS 成人發現有主動脈根部擴大,由心臟超音波檢查常發現伴隨主動脈迴流發生,MFS 婦女懷孕和產後也是主動脈剝離及破裂的高危險時期7,由於動脈壁應力增加與高血液動力循環或激素對主動脈壁的影響而使風險增加。
在骨骼方面 MFS 病人常有前胸畸形 (凸胸或凹胸)、胸椎側彎、長骨過長(肢體細長症)、關節鬆弛及比基因遺傳背景預測的身高還要高的身高 (非必要條件),其他包括蜘蛛狀指、足部外翻、脊柱側彎及自發性氣胸等。
眼睛水晶體異位發生在60-70%的 MFS 患者,原因可能是睫狀體小帶失敗的支撐,其他包括睫狀肌與虹膜發育不全、嚴重近視、早期白內障、青光眼及視網膜剝離等,其他系統包括硬腦膜擴大、皮膚出現萎縮紋或疤痕樣病變及腹股溝疝氣等5。
肆、治療
一、藥物治療
藥物使用主要考量病人主動脈根部擴大的風險,例如相關突變引起主動脈疾病的家族史,MFS 病人除預防性的藥物治療之外,若執行主動脈置換手術,術後應長期服用藥物預防復發。
(一)β-blockers
研究指出 β-blocker 減少心肌收縮、脈搏壓力及改善主動脈彈性,可以降低MFS 近端主動脈的血液動力學壓力,其中 propranolol 是第一個使用於減緩主動脈擴張速度及降低主動脈剝離風險的藥物,病人有較高的存活率8,短期和長期使用 β-blocker 均可改善輕度擴張或更輕度患者的主動脈彈性,雖然 propranolol 是減緩主動脈擴大的首選藥物,但也可以使用其他長效β-blocker 藥物,例如 atenolol 或 metoprolol,其中 atenolol 具有長半衰期、較佳心臟選擇性及較少中樞神經作用和副作用,常被當作首選藥物。
因為 atenolol 可能會傷害胎兒的發展,所以懷孕婦女可選用 labetalol 或 metoprolol,若有主動脈剝離風險之孕婦需控制血壓,應避免使用 nitroprusside,其可能產生的硫氰酸鹽毒性 (thiocyanate toxicity),對胎兒有不利的影響。研究支持 MFS 兒童使用β-blocker,雖然兒童的研究資料還不充分。
2010年美國心臟病學會/美國心臟協會/美國胸腔外科協會 (ACC/AHA/AATS) 胸主動脈指引建議,MFS 成人及主動脈瘤使用 β-blocker 減少主動脈擴張,除非有使用禁忌9,例如心臟傳導阻滯或氣喘等,β-blocker 的劑量應該調整到次極量運動 (在兩個階梯跑上跑下運動) 之後心跳率成人能維持在每分鐘 < 100下,兒童每分鐘 < 110下。
若病人無法耐受 β-blocker 則改用ARB (研究指出可選擇 losartan) 或 ACEI 可能有好處,而 verapamil 也可以當作二線藥物,研究顯示,這些藥物可以使主動脈生長速度輕微下降。對於進行主動脈瓣置換患者,術後需終身服用 β-blocker,建議併用 ARB。
(二)作用於renin-angiotensin system (RAS) 藥物
包括 angiotensin converting enzyme inhibitor (ACEI) 或 angiotensin receptor blocker (ARB),藥理作用是經由阻斷在 RAS 的 TGF-β (transforming growth factor-beta) 訊號以減弱 MFS 的臨床表現,這在人類臨床研究及動物實驗中得到證實10,包括阻止病理性主動脈根部的生長,使主動脈壁厚度和結構正常化。實驗用 TGF-β 抗體治療 MFS 突變老鼠,發現可預防心臟瓣膜及主動脈瘤的發展。在人類的研究結果,包括單用 perindopril 或 losartan 或合併使用 β-blocker,都顯示可減緩 MFS 患者主動脈根部擴大的進展,2010年 ACC/AHA/AATS 指引包括建議使用 ARB 是合理的。對於有胸主動脈瘤的病人,在病人能夠忍受較低血壓情況下,使用 β-blocker 再加上 ARB 或 ACEI 是合理的。
(三)Calcium channel blocker (CCB)
有限的小鼠研究報告使用 CCB 出現動脈瘤擴大、破裂及過早死亡,可能增加主動脈併發症的風險,因此避免使用11。
(四)Statins
動物實驗證明 statins 可能減少主動脈中血管平滑肌細胞產生過量的蛋白質,減緩 MFS 小鼠的主動脈根部擴大,但人類還需更多研究12。
(五)抗凝血劑與抗生素治療
對於 MFS 患者進行主動脈瓣或二尖瓣置換,或以前曾有過心內膜炎,給予預防性抗生素仍然有爭議,需個別化的考量,依據美國與歐洲治療指引建議,這些患者給予預防性抗生素是合理的13,14。接受機械瓣膜置換患者需終身服用抗凝血藥物,另外接受生物瓣膜或瓣膜保留手術患者,術後需服用三個月的抗凝血劑,除了給予 warfarin 外,通常再加上低劑量的 aspirin。
二、手術治療
主動脈弓及降主動脈容易發生剝離及動脈瘤,2010年 ACC/AHA/AATS 指引建議,對於 MFS 患者主動脈外徑 ≧ 50 mm,應該進行選擇性手術以避免急性剝離或破裂,而外徑 < 50 mm 者,包括快速擴大 (> 5 mm/年) 及直徑小於50 mm 而有主動脈剝離或進行性主動脈回流的家族史,是手術修補的適應症3,9,主動脈根部擴張相關症狀進展,可選擇預防性主動脈根部置換,對於嚴重主動脈瓣或二尖瓣閉鎖不全,可進行瓣膜重建或置換手術,骨骼系統相關的脊椎側彎、反覆發作氣胸或胸骨畸形等,需考慮手術矯正,其他如眼科症狀包括水晶體異位、青光眼、視網膜剝離及白內障等需定期追蹤與矯正。MFS 婦女妊娠期發生主動脈剝離的風險高須嚴格控制血壓,孕前選擇預防性主動脈根部置換可以使死亡風險降低7。
伍、預防與監測
美國心臟科學會建議 MFS 患者應限制劇烈的運動,一般原則是選擇低到中等強度的運動並建議個人化的評估3,如果父母有 MFS,則子女中發生 MFS 的風險為50%3,因此遺傳諮詢及預防併發症是很重要的。
美國心臟科醫學會對於胸腔主動脈疾病治療指引建議,MFS 成人如果主動脈直徑在一段時間內穩定的話,主動脈直徑小於45 mm 可以一年一次做影像學檢查,如果主動脈直徑 ≧ 45 mm 或一段時間內顯示有意義的變大,建議每年檢查兩次,如果主動脈直徑顯示快速變化(≧ 0.5 cm/year) 或者影響到心臟或瓣膜功能時,建議應該更頻繁的影像學檢查10。
陸、結論
估計有90%以上典型 MFS 患者終其一生會發生相關血管疾病事件,因此給患者帶來不少壓力,在1993年研究 MFS 患者平均壽命為72歲15,由於藥物治療、早期診斷、非侵入性主動脈影像監測和精確的外科技術,大幅改善 MFS 患者的預後及提高生活品質。
表一 與 MFS 類似症狀之基因異常疾病3,9
表二 2010年修訂 Ghent 疾病分類表準則4,6

表三 系統評分 (Systemic score) 準則4,6:(修訂 Ghent 疾病分類表)

Drug Therapy for Marfan Syndrome
Wen-Hwang Chen1, Min-Si Li2
Pharmacy Department of Tungs, Taichung
MetroHarbor Hospital1
Critical Care Department of Tungs, Taichung
MetroHarbor Hospital2
Abstract
Marfan syndrome (MFS) is an abnormal connective tissue disease and the etiology of which is related to the abnormality of fibrillin-1 gene on chromosome 15. The clinical manifestations and severity of the disease varies. The disease involved multiple organ systems and mostly cardiovascular and musculoskeletal systems. The most serious complications involve the heart valves and aorta especially aortic aneurysm and aortic dissection. It is recommended to regularly track the progress of the disease and the timely surgical intervention to repair the heart valve and especially the aorta. Since there is no cure for marfan syndrome, the goal of drug therapy is to slow the progress the disease especially aortic aneurysm and aortic dissection which both are life-threatening risks to the patient.
參考資料:
1. Dieta HC, Cutting GR, Pyeritz RE, et al: Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991 Jul 25;352(6333):337-9.
2. Hilhorst-Hofstee Y, Rijlaarsdam ME, Scholte AJ, et al: The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat 2010; 31:E1915.
3. Alexander Doyle, Jefferson J. Doyle and Harry C. Dietz: Marfan Syndrome. Nelson Texbook of Pedoatrocs, Chapter 702, 3384-3389.
4. Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476-485.
5. RadonicT,de Witte P, Groennk M, et al: Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet 2011; 80:346.
6. https://www.marfan.org/dx/zscore
7. Elkayam U, Ostrzega E, Shotan A, et al: Cardiovascular problems in pregnant women with the Marfan syndrome. AA Intern Med 1995; 123:117.
8. Tsipouras P, Del Mastro R, Sarfarazi M, et al: Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital contractural arachnodactyly to the fibrillin genes on chromosomes 15 and 5. The International Marfan Syndrome Collaborative Study. N Engl J Med 1992; 326:905.
9. Hiratzka LF, Bakris GL, Beckman JA, et al: 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010; 121:e266.
10. Habashi JP, Judge DP , Holm TM, et al: Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312:117.
11. DoylenJJ, Doyle AJ, Wilson NK, et al: A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome.Elife 2015; 4.
12. McLoughlin D, McGuinness J, Byrne J, et al:
Pravastatin reduces Marfan aortic dilation.
Circulation 2011; 124:S168.
13. Nishimura RA, Otto CM, Bonow RO, et al: 2014 AHA/ACC guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:e57.
14. Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC), European Association for Cardio-Thoracic Surgery (EACTS), Vahanian A et al: Guidelines on the management of valvular heart disease (version 2012). Eur Heart J 2012; 33:2451.
15. Silverman DI, Burton KJ, Gray J, et al: Life expectancy in the Marfan syndrome. Am J Cardiol 1995; 75:157.
通訊作者:陳文皇/通訊地址:台中市梧棲區臺灣大道八段699號
服務單位:童綜合醫院藥劑部藥師/聯絡電話:(O) 04-26581919 ext 4651

