淺談體染色體顯性多囊性腎臟病
吳秋玫、黃微瑄、蕭惠娟
高雄市立大同醫院藥學科
摘要
體染色體顯性多囊性腎臟病(autosomal dominant polycystic kidney disease, ADPKD)是常見的遺傳性腎臟疾病,大多數罹患ADPKD的患者最終需要腎臟替代治療。多囊性腎臟病(PKD)是一種多重器官疾病,導致充滿液體囊泡形成在腎臟和其他器官;因囊泡不斷地增加而取代腎實質部分,最終導致腎衰竭。我們回顧體染色體顯性多囊性腎臟病之病理生理學和臨床藥物治療的最新進展。
關鍵字: 體染色體顯性多囊性腎臟病、tolvaptan
壹、前言
ADPKD是常見的腎臟遺傳性疾病,無論在美國或全球皆位居於第四位造成腎臟衰竭主要原因之一。根據流行病學研究顯示,每500-1000人中就有1位罹患ADPKD。在美國約有60萬患者,而全球約有1200萬人被診斷ADPKD。每10位ADPKD患者就有7人進展為腎衰竭,開始進行透析,平均中位數年齡為58歲。男性進展為腎衰竭比率高於女性。在台灣,逾7萬洗腎病人中,PKD病人約佔5-10%,洗腎病人中約有2.4%為ADPKD1,2,3,4。
貳、遺傳和病理生理學
ADPKD致病基因有兩型:86%至96%的病例中,是由第16對染色體上的PKD1基因突變引起,該基因編碼蛋白polycystin 1。其餘是由第4對染色體上的PKD2基因突變引起的,該基因編碼蛋白polycystin 2。另有一些家族性病例與這兩個基因位均無關。Polycystin 1調節腎小管上皮細胞的黏附和分化;polycystin 2其功能為鈣離子通道,突變會導致液體分泌至囊泡中。這些蛋白突變可能改變腎臟細胞纖毛的功能,影響腎小管感應液體的流動,推測腎小管細胞的增生和分化與流速有關,而纖毛功能受損可能導致囊泡變性。囊泡形成的機制可能為:1.腎小管上皮細胞異常的增生。2.上皮細胞液體分泌。3.細胞外基質異常。4.細胞內鈣離子、cyclic AMP (adenosine monophosphate)、mTOR分子(mammalian target of rapamycin)影響細胞增生與液體分泌。5.血管生成導致囊泡生長1,2,5。
疾病早期,腎小管擴張並慢慢充滿腎絲球濾液。最終,腎小管脫離正常功能的腎元,並充滿分泌液而非過濾液,形成囊泡。可能發生囊泡出血造成血尿。病人罹患急性腎盂腎炎、囊泡感染和尿路結石的風險也較高。除了腎臟的特徵外,可能出現其他器官的表現如:肝臟囊泡、胰臟和腸囊泡、結腸憩室、腹股溝和腹壁疝氣、心臟瓣膜疾病(常見二尖瓣脫垂和主動脈瓣關閉不全)、動脈瘤6。
參、臨床症狀
ADPKD通常最初不會引起任何症狀。一半的患者保持無症狀,未發展為腎功能不全或衰竭,也未被診斷出來。大多數出現症狀的患者會在20多歲時出現症狀。症狀包括由於囊泡腫大和感染症狀引起的腰部、腹部和下背疼痛。發生急性疼痛時,通常是由於囊泡出血或結石通過引起的。發燒常見由急性腎盂腎炎引起的,囊泡破裂進入腹膜後間隙可能引起發燒,持續數週。如果肝囊腫增大或被感染,可能會引起右上腹疼痛。腦部動脈瘤未破裂可能無症狀或可能包括頭痛、噁心和嘔吐以及顱神經功能缺損,這些表現需要立即處理。一些非特異性症狀包括血尿、高血壓、蛋白尿1,2。
肆、診斷和預後
ADPKD一般根據影像學檢查(腎臟超音波、電腦斷層掃描、核磁共振造影發現囊泡)作診斷。基於超音波檢查的成本低、安全性和可用性,是確認疑似ADPKD診斷的影像學檢查首選。診斷需依據家族史和不同年齡的腎臟囊泡數目來定義;超音波診斷標準為:15-39歲患者單側或雙側腎泡數目≥3;40-59歲每側腎臟囊腫數目≥2;60歲以上患者每側腎臟囊腫數≥4,依年齡大小的腎臟囊泡數目且家族史陽性的患者可診斷為ADPKD1。基因檢測可輔助診斷,但尚未常規使用。
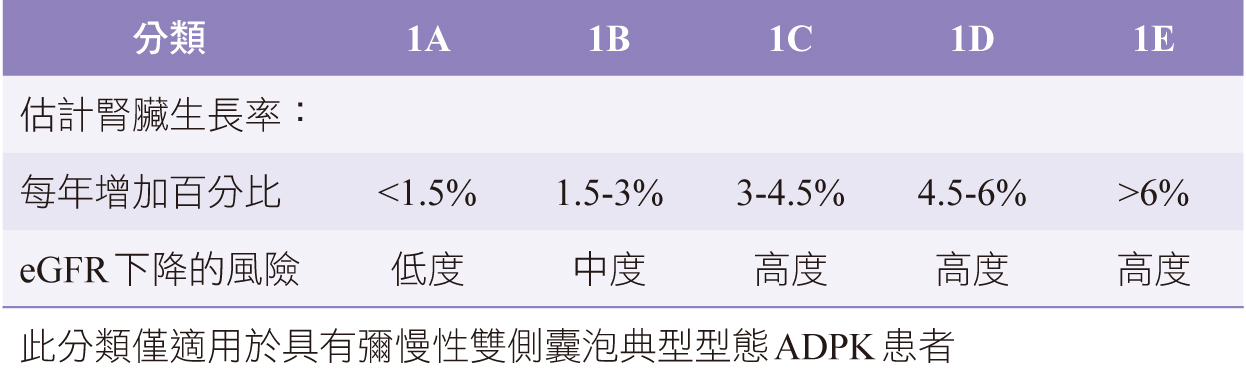
ADPKD實驗室監測包括血清肌酸酐和估計的腎絲球過濾率(estimated Glomerular Filtration Rate, eGFR)值。但疾病進展早期由於eGFR數值偏高會遮蔽腎實質的損傷,且觀察到血清肌酸酐通常在疾病開始3-5年內不會增加。而整體腎臟體積(total kidney volume, TKV)於早期ADPKD且腎功能良好的病人會隨著腎功能下降而增加。TKV已被證實是ADPKD臨床試驗中有用的生物標誌,並且對疾病進展的敏感性高於GFR或血清肌酸酐。在美國Consortium for Radiologic Imaging Studies in Polycystic Kidney Disease (CRISP)已證實腎臟大小的增加是PKD的最早表現;TKV的增加與囊泡體積的增加和GFR的降低相關。CRISP研究中發現,較高的TKV與蛋白尿,微量白蛋白尿,高血壓,嚴重血尿和腎臟功能逐漸喪失有關。導致ADPKD快速進展的危險因子因素整理如(表一)1。Mayo分類系統是定義高風險ADPKD,透過病人年齡、身高和TKV計算,能準確預測ADPKD患者每年腎臟體積增大的速度與腎功能下降的風險而發展為晚期腎臟病(表二)7。
表一 導致ADPKD快速進展之危險因子

表二 Mayo分類系統
伍、治療
一、所有ADPKD患者的治療
(一) 控制血壓
在沒有禁忌症的情況下,通常建議使用血管收縮素轉化酶抑制劑(angiotensin converting enzyme inhibitor, ACEI)或血管收縮素受體阻斷劑(angiotensin receptor blocker, ARB),針對健康的個體年齡在18至50歲,eGFR> 60 mL / min / 1.73 m2,血壓控制目標能達到<110/75 mmHg。 在這樣的患者,積極的控制血壓可能會降低腎臟的生長速度,而在高危患者(Mayo 1D和1E)中,可能會減慢eGFR的下降速度。而其他患者中,使用ACEI或ARB進行治療,使目標血壓控制<130/80 mmHg。
(二) 飲食中的鈉鹽限制
建議所有PKD患者都限制飲食中鈉的攝取,目標是建議2000 毫克/天或更少(換算成每日可使用的鹽量約為5000毫克)。Halt Progression of Polycystic Kidney Disease (HALT-PKD)試驗,所有參與者每天將飲食中的鈉含量限制在2400毫克以下,結果顯示較高的鈉排泄與腎臟生長和eGFR下降的風險增加有關。減少鈉的攝取除了對eGFR有正向作用外,有助於改善ADPKD患者的血壓控制。
(三) 增加液體攝取量
建議ADPKD患者每天喝> 3公升的液體,除非eGFR <30 mL / min / 1.73 m2或有低鈉血症的風險(例如,服用thiazide利尿劑)。較高的液體攝取有助於減輕ADPKD罹患腎結石的風險,且可以透過抑制血漿加壓素抑制囊泡的生長。
二、針對高風險患者的治療
(一) Tolvaptan
Tolvaptan是高度選擇性的血管加壓素V2受體(vasopressin V2-receptor, V2R)拮抗劑,核准用於治療ADPKD。Tolvaptan 是一種抗利尿激素 (antidiuretic hormone, ADH;又名 arginine vasopressin, AVP)的受體抗拮劑。ADPKD的病人身上,因AVP會作用在遠端腎小管與集尿管的V2受體,使細胞內cAMP增加而刺激細胞增生與囊泡形成。Tolvaptan 的作用則透過結合並抑制V2受體,減少cAMP的產生,而減緩腎功能的惡化、減少腎臟囊泡的增生和感染的機會。
Tolvaptan建議劑量從早上45mg/下午15mg開始,耐受性良好可每1-4週調整劑量,最大劑量為早上90mg/下午30mg。早上與下午時間至少間隔8小時且下午服藥時間建議於4點前完成,以減少嚴重的夜尿情形。
在TEMPO(Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes)試驗中,包括1157例eGFR> 60 mL / min且TKV> 750 mL的患者,每年TKV的增加與安慰劑相比,接受tolvaptan治療者TKV增加減少(分別為2.8%和5.5%)。與安慰劑相比,接受tolvaptan治療者每年eGFR下降速度較慢(-2.72對-3.70 mL / min / 1.73 m2 /年)。研究結果也顯示Tolvaptan降低了三年內腎功能的下降率(hazard ratio [HR] 0.39,95% CI 0.26-0.57),及臨床上顯著的腎臟疼痛率(HR 0.64,95% CI 0.47-0.89)8,9。
在REPRISE(Replicating Evidence of Preserved Renal Function: An Investigation of Tolvaptan Safety and Efficacy in ADPKD)試驗中,研究了tolvaptan對eGFR降低的ADPKD患者之影響。收入eGFR 25-65 mL / min / 1.73 m2、年齡18至55歲和eGFR 25-44 mL / min / 1.73 m2、年齡56至65歲患者。1370例患者經12個月tolvaptan治療結果顯示eGFR變化低於安慰劑組(-2.34 vs -3.61 mL / min / 1.73m2)8,9。
安全性方面,口渴、多尿為較常見的藥物副作用,可能會導致病人無法耐受較高劑量的Tolvaptan;另外,肝臟指數上升、尿酸上升也是需要定期監測的可能副作用8,9。衛生福利部在去年10月已經正式給付此藥物用於已出現病情迅速惡化跡象之第3期慢性腎臟病18-50歲自體顯性ADPKD患者,且腎臟影像呈雙側/瀰漫性水泡,病情須符合下列至少一項:(1)一年之內eGFR下降≧5.0 mL/min/1.73 m2 或五年內eGFR每年下降≧2.5mL/min/1.73 m2,且排除其它如脫水、藥物、感染、阻塞等原因所致。(2)身高調整後的整體腎臟體積(height-adjusted total kidney volume, htTKV)符合Mayo分期1C-1E disease。
(二) Octreotide
Octreotide是長效型Somatostatin analogue,可透過結合其受體抑制cAMP形成,減少細胞增生與囊泡形成。一個隨機雙盲安慰劑對照組的臨床試驗,Octreotide相較於安慰劑,一年治療明顯減少TKV(+0.25 vs +8.6%),但腎功能的下降在兩組並未有差異(-5.1 vs -7.2%)10。
(三) Sirolimus、Everolimus
Sirolimus與everolimus是mammalian target of rapamycin (mTOR) inhibitor,透過抑制mTOR減少細胞增生與囊泡形成。一個sirolimus為期18個月,收入100例ADPKD患者,隨機分配接受everolimus或安慰劑,結果顯示無法看到everolimus對TKV與eGFR之差異性11。一個everolimus為期2年的雙盲試驗,收入433例ADPKD患者,隨機分配接受everolimus或安慰劑,結果顯示everolimus減緩了ADPKD患者第一年TKV的增加 (102 vs 157ml; P=0.02),但並未減慢腎功能不全的進展(-8.9 vs -7.7 mL / min / 1.73 m2, P=0.15)12。
陸、結論
新的理論和臨床研究的突破正在闡明ADPKD的病理生理學、自然病程和臨床應用。Tolvaptan是目前唯一核准治療ADPKD藥物,儘管使用時應小心評估其益處和風險,對於某些病人能盡早使用tolvaptan能延長無透析的時間,減緩病程的進展。但並非每位病人都適用tolvaptan,未來仍需更多的臨床研究來找出藥物使用的準則。
Autosomal dominant polycystic kidney disease
Chiou-Mei Wu, Wei-Hsuan Huang, Hsiao Hui-Chuan
Department of Pharmacy, Kaohsiung Municipal
Ta-Tung Hospital,
Kaohsiung Medical University Hospital
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a common hereditary kidney disease. Most patients suffering from ADPKD will eventually need renal replacement therapy. Polycystic kidney disease (PKD) is a multiorgan disorder that causes fluid-filled vesicles to form in the kidneys and other organs; the vesicles continue to increase and replace the renal parenchyma, eventually leading to kidney failure. We review the pathophysiology and pharmacologic treatment of autosomal dominant polycystic kidney disease.
參考資料:
1. Nobakht N, Hanna RM, Al-Baghdadi M, et al: Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review. Kidney Med. 2020;2(2):196-208.
2. Torres VE, Bennett WM. (2021). Autosomal dominant polycystic kidney disease (ADPKD) in adults: Epidemiology, clinical presentation, and diagnosis. UpToDate. Retrieved February 22, 2021, from https://www.uptodate.com/contents/autosomal-dominant-polycystic-kidney-disease-adpkd-in-adults-epidemiology-clinical-presentation-and-diagnosis
3. Lee LJ, Kao TW, Chu TS. Epidemiology of Polycystic Kidney Disease in Taiwan. Formosan J Med. 2017;21:427-433.
4. Bergmann C, Guay-Woodford LM, Harris PC, et al: Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50
5. Pei Y, Watnick F. (2020). Autosomal dominant polycystic kidney disease (ADPKD): Genetics of the disease and mechanisms of cyst growth. UpToDate. Retrieved February 22, 2021, from https://www.uptodate.com/contents/autosomal-dominant-polycystic-kidney-disease-adpkd-genetics-of-the-disease-and-mechanisms-of-cyst-growth
6. Bennett WM, Torres VE. (2020). Autosomal dominant polycystic kidney disease (ADPKD): Extrarenal manifestations. UpToDate. Retrieved February 22, 2021, from https://www.uptodate.com/contents/autosomal-dominant-polycystic-kidney-disease-adpkd-extrarenal-manifestations
7. Irazabal MV, Rangel LJ, Bergstralh EJ, et al: Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015; 26(1):160-172.
8. Chapman AB, Rahbari-Oskoui FF, Bennett WM. (2020). Autosomal dominant polycystic kidney disease (ADPKD): Treatment. UpToDate. Retrieved February 22, 2021, from https://www.uptodate.com/contents/autosomal-dominant-polycystic-kidney-disease-adpkd-treatment
9. Blair HA. Tolvaptan: A Review in Autosomal Dominant Polycystic Kidney Disease. Drugs. 2019;79(3):303-313.
10. Griffiths J, Mills MT, Ong AC. Long-acting somatostatin analogue treatments in autosomal dominant polycystic kidney disease and polycystic liver disease: a systematic review and meta-analysis. BMJ Open. 2020;10(1):e032620
11. Serra AL, Poster D, Kistler AD, et al: Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. Engl J Med. 2010;363(9):820-829.
12. Walz G, Budde K, Mannaa M, et al: Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):830-840.
通訊作者:蕭惠娟/通訊地址:高雄市前金區中華三路68號
服務單位:高雄市立大同醫院藥學科/聯絡電話:(O) 07-2911101 ext 8378

